










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkSr. Director:
Las lipodistrofias son un grupo heterogéneo de enfermedades muy poco frecuentes, caracterizadas por la pérdida selectiva de tejido adiposo, lo que condiciona, frecuentemente, una insulinorresistencia con tendencia al desarrollo de: diabetes, dislipemia, esteatosis hepática, acantosis nigricans e hiperandrogenismo1. La lipodistrofia parcial adquirida (LPA; OMIM: 608709), también conocida como síndrome de Barraquer-Simons, es una de las más frecuentes y afecta preferentemente a mujeres (4:1). Se caracteriza por una pérdida progresiva del tejido graso subcutáneo, de inicio habitualmente en la infancia2, y que progresa de forma cefalocaudal, afectando a: cara, extremidades superiores, tronco y abdomen. A diferencia de lo que ocurre en otras lipodistrofias, la insulinorresistencia y las complicaciones metabólicas, salvo la hepatomegalia (60%), son muy infrecuentes1,2.
La LPA, en un 10-20% de los casos, se asocia a enfermedades autoinmunes, especialmente al lupus, pero su complicación más habitual (20%) es el desarrollo, una media de 8 años después del inicio de la enfermedad, de una glomerulonefritis membranoproliferativa tipo 2 o enfermedad de depósitos densos (DDD), en cuya patogenia está implicada la activación de la vía alternativa del complemento (VAC)3. Los pacientes muestran niveles séricos disminuidos de C3 (70%) y positividad para C3NeF (80%)2, un autoanticuerpo capaz de alterar la VAC y para el que, en el caso de la LPA, se ha postulado también un potencial papel en la destrucción del tejido graso4.
Niña de 15 años de edad, de etnia caucásica y sin antecedentes familiares ni personales de interés, diagnosticada de LPA, por pérdida progresiva, desde los 5 años, de la grasa subcutánea en cara (figs. 1a y b), con posterior afectación de cuello y hombros. En sus revisiones periódicas, no se habían observado alteraciones clínicas ni analíticas, salvo la lenta progresión de la lipoatrofia y la presencia mantenida de niveles séricos disminuidos de C3 (26-48 mg/dl; VN: 86-184). Nunca había presentado datos clínicos ni analíticos sugerentes de nefropatía, dislipemia, insulinorresistencia o hiperandrogenismo. Los niveles séricos de leptina (8,03 ng/ml; VN: 15,3 ± 8,1 DE) y de adiponectina (8,3 mg/ml; VN: 12,0 ± 3,1 DE) se encontraron ligeramente disminuidos y el estudio de composición corporal por DXA mostró una disminución de la grasa corporal total (17,7%; VN: 25,9 ± 6,3).
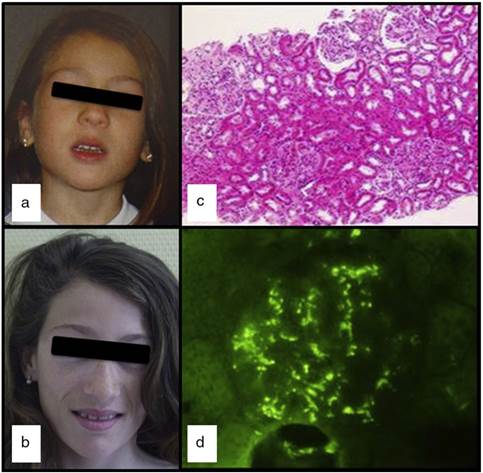
Figura 1 a y b) Evolución de la pérdida de grasa en la cara; c) Glomérulos con aumento de la celularidad mesangial, y afectación focal y segmentaria (H&E × 100) y d) Inmunofluorescencia granular mesangial con anti-IgA.
A los 13 años de edad, tras 3 días de fiebre y en el contexto de una gastroenteritis aguda, presenta hematuria macroscópica con proteinuria no nefrótica (13 mg/kg/día) y elevación transitoria de la creatinina (máximo: 0,74 mg/dl). La fiebre desapareció 24 h después, pero la hematuria macroscópica persistió durante 2 semanas, sin otra sintomatología y con mejoría de la función renal. Los niveles séricos de IgA se encontraron elevados (377 mg/dl; VN: 40-350), el C3 se mantenía bajo y el C3NeF negativo. Los estudios de autoinmunidad (anticuerpos antitiroideos, antineutrófilos y antinucleares) fueron negativos. La biopsia renal percutánea, realizada un mes después de resuelta la hematuria, contenía 14 glomérulos y reveló la presencia de hipercelularidad mesangial focal y segmentaria (fig. 1c): M1, E0, S0 y T0, según la clasificación de Oxford5 (hipercelularidad mesangial [M], proliferación endocapilar [E] glomeruloesclerosis segmentaria [S] y fibrosis intersticial con atrofia tubular [T]). Solo uno de los glomérulos presentaba una semiluna que ocupaba el 26-50% del glomérulo. La inmunofluorescencia mostró depósitos granulares mesangiales de IgA y C3 (fig. 1d). La microscopia electrónica confirmó la presencia de depósitos electrodensos en el glomérulo, descartando DDD. La paciente fue diagnosticada de nefropatía IgA (NIgA), alcanzando la remisión clínica, con desaparición espontánea de la proteinuria y hematuria, pero manteniendo niveles séricos disminuidos de C3 (33 mg/dl) y elevados de IgA (353 mg/dl).
La LPA es una enfermedad infrecuente y de etiopatogenia incierta. Su asociación a enfermedades autoinmunes y a DDD, así como la disminución de C3 y la positividad de C3NeF sugieren una base autoinmune2; aunque, se ha postulado, también, la existencia de una predisposición de base genética6. La NIgA, por su parte, es la glomerulopatía más frecuente en el mundo7. En su etiopatogenia, tampoco plenamente aclarada, está implicada también la activación del complemento a través de la VAC7-9 y, en ocasiones, a través de la vía de la lectina10. El diagnóstico es histológico y la biopsia renal muestra inmunodepósitos mesangiales de IgA1 con C3 y, ocasionalmente, IgG o IgM. En los depósitos mesangiales, es frecuente el hallazgo de componentes de la VAC (C3 y properdina), pero no de la vía clásica (C1q y C4), lo que unido a la normalidad sérica de C3 y de otros componentes del complemento sugiere que la activación del complemento se produciría en el propio riñón.
En resumen, presentamos el primer caso descrito de asociación de LPA y NIgA. Aunque, dada la incidencia de la NIgA, podría tratarse de una coincidencia, el hecho de que la LPA asocie con frecuencia una nefropatía, la DDD, en cuya patogenia, al igual que ocurre en la NIgA, está implicada la activación de la VAC, plantea la posibilidad de un nexo común entre ambas enfermedades.

