










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
La Neurofibromatosis tipo I (NF-1) o enfermedad de von Recklinghausen es una enfermedad genética autosómica dominante, aunque hasta el 50% de los casos puede darse de manera esporádica, que ocurre en aproximadamente uno de cada 3.000 nacidos vivos. Está causada por una alteración en el gen Nf-1 del cromosoma 17 que codifica la neurofibromina, proteína responsable de mecanismos de control encargados de la regulación de la división celular1. El diagnóstico de esta patología es clínico: se presenta con neurofibromas en cualquier localización corporal, manchas color café con leche, efélides en axilas e ingles, o neoplasias a nivel del sistema nervioso central (frecuentemente gliomas de nervio óptico), y los pacientes afectados presentan con frecuencia pequeños hamartomas del iris (nódulos de Lisch) o lesiones displásicas óseas. La afectación renal secundaria en estos pacientes es rara y, aunque se han descrito algunos casos de enfermedad glomerular, la asociación a patología vascular renal y la predisposición al desarrollo de tumores neuroendocrinos a nivel suprarrenal (feocromocitomas), por mecanismos no claramente explicados hasta el momento2-3, han sido las observaciones más frecuentes
Presentamos dos casos de síndrome nefrótico en pacientes con diagnóstico de NF-1
Caso clínico 1
Mujer de 41 años con antecedentes de dislipemia en tratamiento con estatinas y manchas cutáneas tipo café con leche desde el nacimiento al igual que su padre y hermano (Fig. 1A). Fue diagnosticada de enfermedad de cambios mínimos por la demostración de fusión de podocitos con inmunofluorescencia negativa mediante biopsia renal (Fig. 2) seis años antes, por un cuadro de síndrome nefrótico (proteinuria de 8 g en 24 horas, sin hematuria) de debut agudo, con función renal y presión arterial normales, tras haberse descartado razonablemente causas tóxicas, neoplasias o infecciones concomitantes y etiología autoinmune (lupus, vasculitis o mieloma múltiple). Recibió tratamiento esteroideo (1 mg/kg/día) durante tres meses, con remisión clínica pero sin remisión completa de la proteinuria (persistencia de 4,1 g en 24 horas)
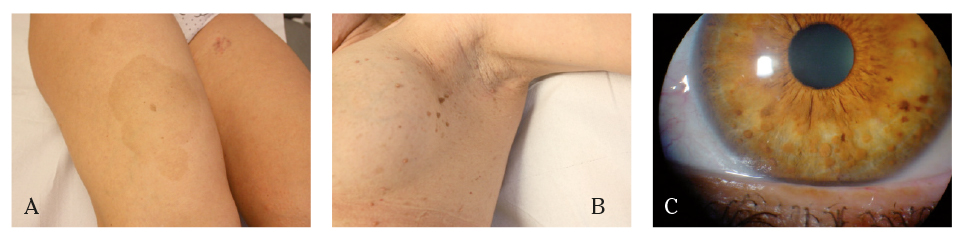
Figura 1. Manifestaciones de la neurofibromatosis tipo I. A. Manchas cutáneas café con leche. B. Neurofibromas en zona axilar C. Nódulos de Lisch
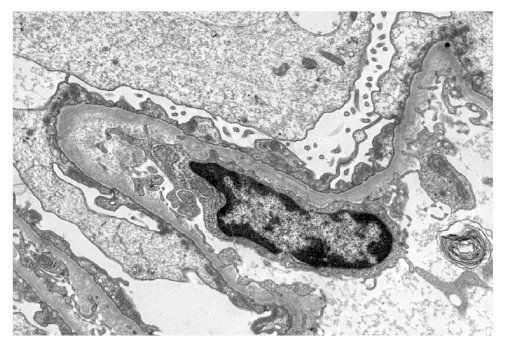
Figura 2. Alteración renal por enfermedad de cambios mínimos: fusión podocitaria con fusión pedicelar y transformación en una gruesa lámina citoplasmática de pedicelos que reviste el polo externo de la membrana basal glomerular. Microscopía electrónica de transmisión, 5.500x
En el seguimiento posterior se observó la aparición de lesiones cutáneas de pequeño tamaño y rápido crecimiento, compatibles con neurofibromas (Fig. 1B), y lesiones en iris no objetivadas previamente (Fig. 1C), altamente sugestivas de NF-1, por lo que se estableció el diagnóstico clínico, sin evidencias de afectación del sistema nervioso central
A los cuatro meses de la suspensión de los esteroides presentó su primera recidiva clínica y un repunte en la proteinuria, recibiendo de nuevo tratamiento esteroideo. Tras dos brotes posteriores en meses siguientes, con respuesta parcial (proteinurias residuales de 2 a 3 g en 24 horas), se consideró a la paciente corticorresistente, por lo que se inició tratamiento con ciclosporina (CsA) en combinación con dosis bajas de esteroides. Sin embargo, en los años sucesivos ha presentado cuatro brotes de síndrome nefrótico sin conseguir la retirada completa de los esteroides
A los tres años del diagnóstico tuvo un embarazo a término, con un niño sano y un aumento importante de la proteinuria hasta 6,5 g en 24 horas, que requirió de nuevo el aumento de la dosis de esteroides. La función renal se ha mantenido normal hasta la actualidad, en que presenta una proteinuria menor de 1 g en 24 horas, en tratamiento con CsA y prednisona 5 mg al día
Caso clínico 2
Mujer de 71 años con antecedente de sarcoma de partes blandas en muslo derecho tratado con radioterapia, cirugía y quimioterapia en el contexto de un diagnóstico clínico previo de NF-1 (presencia de neurofibromas generalizados por todo el cuerpo). Fue valorada en el servicio de Nefrología por síndrome nefrótico junto con insuficiencia renal progresiva. La biopsia renal reveló depósitos amiloideos de proteÍna de tipo AA a nivel glomerular y en los vasos, con confirmación inmunohistoquímica (Fig. 3) y compatible con diagnóstico de glomerulopatía por amiloidosis secundaria; se descartó depósito amiloideo en otros órganos (a nivel cardíaco o neurológico)
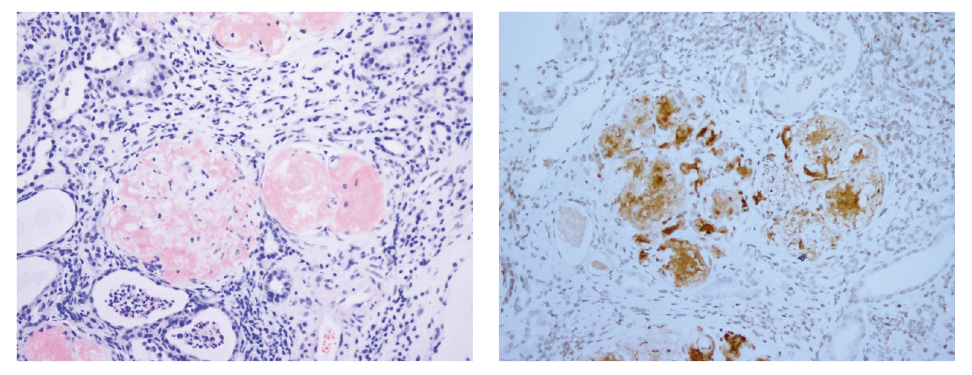
Figura 3. Depósito amiloideo (material hialino acelular) que ocupa glomérulos renales. A. Rojo Congo, 200x. B. Inmunohistoquímica para proteína amiloide AA, 200x
No recibió tratamiento específico y durante el seguimiento posterior la función renal siguió empeorando progresivamente, acompañada en todo momento de proteinuria en rango nefrótico. A los dos años de seguimiento, y en situación de enfermedad renal crónica estadio 4, presentó una masa tumoral a nivel sacro con origen en la vaina del nervio periférico acompañada de destrucción ósea local y metástasis a distancia, que abocó en el fallecimiento de la paciente a pesar del tratamiento con radioterapia
Discusión
La NF-1 es una enfermedad autosómica dominante que resulta de la desregulación de la vía de activación PI3K/AKT/mTOR causada por la mutación en el gen Nf-1 en el cromosoma 17q11.24. La forma más frecuente de afectación renal secundaria a la NF-1 es la vascular, en forma de estenosis de la arteria renal de forma uni o bilateral3,5. Una de las formas de presentación más típica de NF-1 es la hipertensión arterial en niños por afectación de la vascularización renal3. Excepcionalmente, se han descrito casos de compresión de la vena renal secundarios a un neurofibroma6, o a un aneurisma en la arteria renal secundaria a NF-17
Las descripciones de afectación glomerular en la NF-1 son escasas, e incluyen casos aislados de glomerulosclerosis segmentaria y focal (GSFS)8, nefropatía membranosa (NM)5,9-10, nefropatía por IgA11 y enfermedad por cambios mínimos (ECM)12
Presentamos dos casos de síndrome nefrótico en pacientes con diagnóstico de NF-1. La ECM es la causa más frecuente de síndrome nefrótico en niños y una de las más comunes en adultos, cuya característica clínica principal es la fusión podocitaria, visible mediante microscopía electrónica, perdiéndose la integridad de la barrera de filtración glomerular; los mecanismos propuestos incluyen la existencia de algún factor circulante producido por la disfunción de células T13. Las formas secundarias se han asociado a causas farmacológicas, infecciosas, paraneoplásicas, o alérgicas, aunque la NF-1 no se había descrito previamente14. En nuestro caso, el diagnóstico de ECM fue histológico, demostrando la fusión de pedicelos mediante microscopía electrónica con un riñón ópticamente normal e inmunofluorescencia negativa, características que la diferencian de la GSFS. La amiloidosis secundaria, causada por el por depósito extracelular de fragmentos de proteína de tipo amiloide AA, está asociada enfermedades inflamatorias de curso crónico15. La asociación de NF-1 y ECM es anecdótica en la bibliografía consultada16. También son escasas las descripciones de asociación de NF-1 y almiloidosis secundaria; De Schepper y col, en un estudio sobre la etiopatogenia de la pigmentación de la piel en los pacientes con NF-1, describen la interacción entre la proteína precursora de amiloide y la GTPasa activada por el dominio de la proteína transmembrana neurofibromina 117 (codificada por el gen mutado Nf-118)
La neurofibromina 1 se encarga de la regulación de la GTPasa que regula la actividad de las proteínas ras, implicadas en mecanismos de supresión tumoral, inactivando a su vez un tipo de proto-oncogén conocido como p21-ras12. Estas proteínas tipo ras contribuyen al balance homeostático de los podocitos glomerulares. Por este motivo, se ha descrito una regulación aumentada de ras en la NF-1, que podría ser la responsable de inducir la activación de las vías de señalización del factor de crecimiento toti-potencial SCF/c-kit, mTOR y MAPK16, que se ha asociado a una reducción en el número de nefronas y en el desarrollo de glomeruloresclerosis e insuficiencia renal en modelos animales y humanos19-20. La neurofibromina 1 podría ser responsable de la regulación de mTOR por esta vía, y se ha descrito una reducción en el volumen de astrocitomas y de otros tumores asociados en pacientes en tratamiento con everolimus (inhibidor mTOR)4. Ito y col mostraron que la activación del complejo mTOR-1 induce alteración en las propiedades de algunas proteínas, e incluso influye en la localización de la nefrina en el podocito, lo que conllevaría al desarrollo de proteinuria en modelos animales de glomerulonefritis de cambios mínimos21
Existen en la actualidad muy pocas publicaciones de casos de glomerulonefritis secundaria asociada a NF-1, y aunque en nuestros casos la asociación de ambas entidades se hizo por la coexistencia clínica, resulta muy complicado establecer patrones de desarrollo y mecanismos patofisiológicos que definan esta asociación

