










 Servicios personalizados
Servicios personalizados
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCTION
ALDS is globally used for the treatment and/or prevention of postmenopausal osteoporosis and glucocorticoid-induced osteoporosis1. It belongs to BCS III class (high solubility and low permeability). Low oral bioavailability (under 1%) is one of the most important disadvantages of ALDS. That is caused by several factors such as low permeability due to its negatively charged molecules (AL belongs to the 3rd class of biopharmaceutical classification system); short plasma half-time (t½ 0.5-2 h)2 and its chelation by Ca2+ ions which results in non-absorbable complexes.
There is a need to develop more efficient delivery systems of ALDS to avoid the upper gastrointestinal tract irritation3. Symptoms of upper gastrointestinal tract irritation including esophagitis, vomiting, epigastric pain, nausea, and dyspepsia, have been reported with use of ALDS sodium tablets. Esophageal adverse effects have been reported with use of ALDS tablet, some of which were more severe and dangerous than that observed in controlled clinical trials. It is important to note that some cases of esophagitis and also esophageal ulceration were related to ALDS tablet use; many of these cases have occurred in patients who did not follow the recommended instructions or who continued using ALDS in spite of worsening esophageal symptoms. About 50% of patients discontinue taking ALDS tablets during the first year of treatment due to its gastrointestinal tract (GIT) side effects.
Research on ALDS is a challenge as its quantitative determination is sophisticated From the analytical point of view, sodium ALDS assay is a difficult task due to the lack of chromophores absorbing photons between 200 and 800 nm4. One of the most promising delivery systems is liposomes formulations. The liposome is one of the potential drug delivery systems applying nanotechnology to potentiate the therapeutic efficacy and reduce toxicities of conventional medicines, they are consist of lipid vesicles from natural and synthetic phospholipids of different sizes, loading capacities, and compositions. They are defined as vesicular structures consisting of amphiphilic, in vivo degradable phospholipids and can thus encapsulate a broad range of effective agents, that effective agents may have hydrophilic, lipophilic or even amphiphilic nature. There are three main categories of liposomes: small unilamellar vesicles, large unilamellar vesicles, and multilamellar vesicles.
That classification is based on liposomes lamellarity and size. Liposomes have been used as carrier systems for the delivery of vaccines, hormones, and therapeutic drugs, due to the easy preparation, biodegradability, good biocompatibility, and commercial availability of liposomes5. Earlier studies have discovered that liposomal drug delivery systems improved the cellular absorption of highly hydrophilic drugs such as acyclovir and cefotaxime6. Nanoparticles may be comprised of several kind materials being classified as nondegradable and biodegradable7.
Recently, Polysaccharide-based nanoparticles have received high attention as very promising nanoparticulate drug delivery systems due to their unique potentials as inert excipient biopolymer8. The use of starch nanoparticles is, therefore, receiving a significant amount of attention due to their good biocompatibility and biodegradability9. Starch is relatively pure and does not need much purification processes like other naturally- occurring biopolymers e.g. celluloses and gums10.
By optimizing the right combination of liposomal and polymer characteristics, it is possible to develop delivery systems for the specific, prolonged, and controlled release11. High amylose corn starch has high sustained release properties due to its excellent gel-forming capacity12. The mechanism of drug release from such gel-forming matrix assumes a controlled passage of drug molecules through the obstructive gel layer, gel structure, and matrix. When starch granules are heated in the aqueous environment, granules start swelling and gelatinization takes place at about 55- 80°C depending on the type of starch used13. Upon cooling starch, it begins to undergo retro gradation whereby the starch molecules begin to re-associate in an ordered structure. Cross-linked starch nanoparticles were used for drug delivery and Indomethacin was taken as the model drug14. Recently, efforts were made to develop alternative formulations for the oral administration of ALDS, with a goal of decreasing GI adverse events and possibly increasing compliance15.
MATERIALS AND METHODS
Materials
aLDS was a gift from Unipharma Pharmaceutical Industries (Egypt). Soya bean phosphatidylcholine, cholesterol, and 2-mercaptoethanol (2ME) were purchased from Sigma-Aldrich (USA). Starch was kindly supplied by Starch and Glucose Company (Egypt). Chloroform, methyl alcohol, and sodium hydroxide were obtained from El-Nasr Pharmaceutical Co. (Egypt). All other chemicals and reagents used were of pharmaceutical grade.
Methods
Quantitative analysis of ALDS
Double distilled water was used for the preparation of all solutions.
-Preparation of standard stock and test solutions:
The stock solution was prepared by dissolving 10 mg of ALDS sodium in 10 ml of 0.05 M NaOH and the prepared solution is used with 48 hours. The working solution of the derivatizing reagent was prepared by dissolving 10 mg of o-phthalaldehyde (OPA) in 2 ml of 0.05 M NaOH, then 50 μl of 2-mercaptoethanol (2ME) solution were added. This solution was freshly prepared for each experiment.
The total solution volume was completed to 10 ml using 0.05M NaOH. Aliquots (0.0, 0.4, 0.8, 1.2, 1.6, 2 and 2.4 μM) of this solution were transferred to a 2ml vial. 100 µl of the OPA/2ME reagent was added to each vial and then the volumes were completed to 2 ml with 0.05 M NaOH. 1mg/ml ALDS solution was prepared using 0.05 M NaOH as a solvent.
-Spectrofluorometric determination of ALDS:
The reported methods for quantitation of ALDS are all expensive and time-consuming16. The procedure used in this study was based on the reaction of a primary amino group of ALDS ion with o-phthalaldehyde (OPA) in alkaline medium giving a fluorescent adduct according to the following equation17:

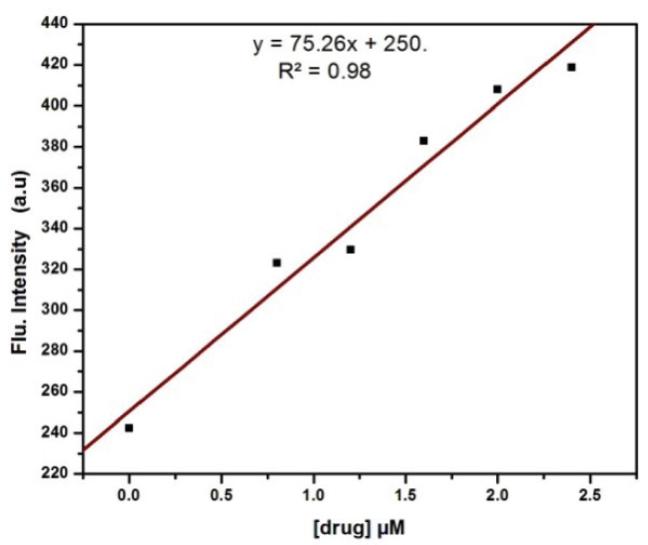
A calibration curve was constructed by plotting fluorescence intensity (in arbitrary units) versus concentration. The linear portion of the curve lies within the concentration range of 0.0-2.4 μM where the inner-filter effect was minimum. The limit of quantification of the method was 29 nanomolar as reported earlier.
-Preparation of calibration curve:
Liposomes were prepared by a modified thin film hydration method18. Phosphatidylcholine (0.6 g) and cholesterol (0.2g) were mixed and then the mixture was dissolved in chloroform/methanol mixture (in a volume ratio 2:3)19. The resulting mixture was dried up to thin lipid films on the inner wall of the flask using a rotary evaporator under vacuum. ALDS (50 mg) was dissolved in 10 ml isotonic NaCl solution, then different amounts of starch were added in incremental amounts (0.0 g to 0.5 g) with proper mixing, and the solution was heated at 55 to 65ºC for 10 to 15 min. The produced lipid thin film was hydrated with the prepared ALDS sodium solution and agitated for a suitable period above 65ºC using rotary evaporator. Samples were sonicated for few minutes and centrifuged at 18000 rpm for 30min. Finally, separation of supernatant has been performed.
Preparation of liposomes
Liposomes were prepared by a modified thin film hydration method18. Phosphatidylcholine (0.6 g) and cholesterol (0.2g) were mixed and then the mixture was dissolved in chloroform/methanol mixture (in a volume ratio 2:3)19. The resulting mixture was dried up to thin lipid films on the inner wall of the flask using a rotary evaporator under vacuum. ALDS (50 mg) was dissolved in 10 ml isotonic NaCl solution, then different amounts of starch were added in incremental amounts (0.0 g to 0.5 g) with proper mixing, and the solution was heated at 55 to 65ºC for 10 to 15 min. The produced lipid thin film was hydrated with the prepared ALDS sodium solution and agitated for a suitable period above 65ºC using rotary evaporator. Samples were sonicated for few minutes and centrifuged at 18000 rpm for 30min. Finally, separation of supernatant has been performed.
Determination of vesicle size
After dilution with water, both vesicles size and size distribution were determined by the dynamic light scattering method using a Malvern Mastersizer(UK).
Zeta potential determination
Zeta potentials of liposomes were determined from electrophoretic mobility of the particles using an instrument model Malvern Zetasizer 2000(Malvern, UK) [27].
Entrapment efficiency (EE) determination
The liposomes were separated from the supernatant then the concentrations of ALDS in isotonic NaCl solutions were determined spectrofluorometrically after derivatization using spectrofluorometry (Shimadzu, Kyoto, Japan) at lmax 470 nm.20 The entrapment efficiency expressed as entrapment percentage was calculated using the following equation:
Entrapment Efficiency % = (Entrapped drug / Total drug) × 100
In vitro drug release study
The drug release studies were carried out in 250 ml beakers containing phosphate buffered saline pH 7.4 as at 37 C, absorption was found to be chiefly in the upper part of the small intestine and hence pH 7.4 is more consistence with the small intestine pH. The beaker content was constantly stirred at 150 rpm with a small magnetic stirrer. Dialysis membrane was taken and one end of the membrane was sealed. After separation of entrapped ALDS liposome dispersion was filled in the dialysis membrane and another end was closed. The dialysis membrane containing the sample was suspended in the medium. Aliquots were withdrawn (5 ml) at selected time intervals (0.5, 1, 2, 3, 4, 6, 8 and 24 h), filtered and the apparatus was immediately replenished with the same quantity of fresh buffer medium. The Aliquots were measured for the amount of the drug by using the previous stated derivatizing agents, all reagents are freshly prepared.
Stability study
The in vitro stability was tested for two prepared liposomes formulations F1 (free starch liposomes) and F6 (liposomes with the highest concentration of starch) by monitoring the particle size and zeta potential for over one month at storage temperatures of 4-8 C and 20-25C. To determine the capability of the prepared formulas to retain the drug within the vesicles throughout the storage period, F1 and F6 were centrifuged and the entrapment efficiency was determined as described earlier in the entrapment efficiency determination section.
Transmission electron microscopy (TEM)
The optimum prepared liposome formula (F6) morphology was visualized by transmission electron microscopy (TEM). The prepared liposomes were diluted with double-distilled water then one drop of the prepared formula was added onto hydrophobic carbon. The samples were air dried for few minutes at room temperature. The specimens were stained with uranyl acetate for 4 min and the excess amount of uranyl acetate was blotted using filter paper. After drying, the liposomes on a hydrophobic carbon grid were directly observed and photographs were captured using Transmission Electron Microscope (JEOL Ltd., Tokyo, Japan).
Ulcerogenicity study
Based on the entrapment efficiency, stability and in vitro drug release studies; F6 was selected to be compared with alendronate solution of the same concentration. The ulcerogenicity study was performed for both of them to evaluate their gastric -ulcerogenic potential. Three groups (A, B and C) of rats were administered each consist of 6 rats, rats weights were ranging between 150 and 200 gm. Group A represented the control group, group B represented F6 receiving rates while group C represented rats which received non-liposomal alendronate solution. Observation of the gastric mucosa for the presence of lesions following oral administration of 20 mg/kg of the tested formulas was used as an indication for the ulcerogenic effects. The rats in group B and C were administered alendronate (20 mg/kg) for 7 days, while group A rats were receiving saline, then all the rats have been killed, the stomachs have been removed, and the extent of tissue damage was evaluated microscopically. Ulcer index was calculated to analysis the effect on the stomach mucosa. The ulcer index represents the total sum of Ulcers numbers, incidence percentage and severity of the resulted ulcers after the oral administration the drug.
Results and discussion
Analytical procedure
ALDS sodium has no characteristic absorption or emission chromophores, so its estimation represents a challenge20 due to the lack of chromophores absorbing photons between 200 and 800 nm. The reactions of OPA with amino compounds in the presence of nucleophilic agents to give isoindole derivatives are used for the analytical determination of primary amines and amino acids.
Calibration curve
Figure 1 shows a calibration curve, based on plotting OPA- ALDS adduct fluorescence intensity versus ALDS concentration. The calibration curve was linear over the concentration range of 0.0-2.4 μM and the limit of quantification of the method was 29 nanomolar that is comparable with the previous studies21.
Vesicles size analysis
Liposomes particle sizes ranged from 94 nm to 298 nm and that range was increasing with elevating starch concentration as illustrated in table 2. This wide size distribution range may indicate liposomes aggregation22.
Table 1 Liposomes formulations
| Formula no. | Phosphatidylcholine (gm( | Starch (g ( | Cholesterol (g ( |
|---|---|---|---|
| 1 | 0.6 | 0 | 0.2 |
| 2 | 0.6 | 0.1 | 0.2 |
| 3 | 0.6 | 0.2 | 0.2 |
| 4 | 0.6 | 0.3 | 0.2 |
| 5 | 0.6 | 0.4 | 0.2 |
| 6 | 0.6 | 0.5 | 0.2 |
Table 2 Particle size distribution of the prepared liposomes
| Formula no. | Starch concentration (%) | Particle size (nm) | Mean polydispersity index(PDI) |
|---|---|---|---|
| 1 | 0 | 94±4.3 | 0.27±0.7 |
| 2 | 0.1 | 191±6.1 | 0.14±0.9 |
| 3 | 0.2 | 225±5.9 | 0.17±0.8 |
| 4 | 0.3 | 242±7.1 | 0.19±0.4 |
| 5 | 0.4 | 278±2.2 | 0.29±1.3 |
| 6 | 0.5 | 298±3.5 | 0.07±0.2 |
It is noticed that with increasing starch concentration there is a relative increase in the size of liposomes that may be attributed to the formation of the starch outer layer around liposomes vesicles that results are comparable with previous studies which investigated the effect of natural polymers incorporation on liposomes physical characters23.
Zeta potential
Zeta potential has frequently been used to characterize colloidal drug delivery systems. It is a measure of the particles surface electrical charge. The zeta potential is a good indicator of the stability of the colloidal system. If all the particles in suspension have a large positive or negative zeta potential, then they will repel each other, and there will be no affinity for the particles to agglomerate. The Zeta potential is a significant parameter which influences liposomal behavior. In vivo, the surface charge density influences the liposomes' distribution, and in vitro, a high potential may contribute to the liposomes physical stability by decreasing the rate of aggregation. ALDS liposomes without starch showed a slight negative zeta potential, in agreement with early reports24.
It was obvious that coating of liposomes by starch shifted the zeta potential values, from slightly negative to larger negative. The results show that liposomes had relatively large zeta values after being coated with 0.1–0.5% w/v starch solutions. The liposomes had a zeta potential ranging from -12 mV to -39 mV as shown in table 3 giving good stability of the liposomes25.
Entrapment efficiency (EE)
Concerning the effect of starch content on the liposome encapsulation efficiency of ALDS, results showed that the percentage entrapment efficiency of ALDS increased by increasing starch content. The percentage entrapment efficiencies of ALDS liposomes were 46.5%, 60.9%, 67.2%, 72.1%, 76.3% and 78.5% for the starch concentration ratios of 0 %, 0.1%, 0.2%,0.3%,0.4%, and 0.5% respectively as shown in Table 4.
Table 4 Percentage entrapment efficiency (EE%) of ALDS by liposomes of different starch concentrations
| Liposomal formula | Entrapment efficiency (% w/w) | ± SD |
|---|---|---|
| 1 | 46.5 | ±1.89 |
| 2 | 60.9 | ±3.05 |
| 3 | 67.2 | ±1.63 |
| 4 | 72.1 | ±1.89 |
| 5 | 76.3 | ±1.51 |
| 6 | 78.5 | ±1.32 |
EE% is used to evaluate the effect of using different concentrations of starch during formulation. According to previous results, upon increasing the starch content, the nanoliposomes become more stable and the permeability decreases. This leads to more drug retention due to drug entrapment through the obstructive gel matrix26. The same results were obtained when other natural polymers such as chitosan are used in the liposome preparation.
The In vitro release study
The drug release study from the six nano-liposomal formulations was studied and compared to the pure drug as shown in (Fig. no. 2). Liposomal formulation exhibit slow release that may be attributed to the rigidity of the liposome membrane which will increase permeability barrier and so reduce the drug loss rate. Moreover, It was found that the formulation F1 which is starch free has faster release comparing with the formulation that contains starch that maybe due to the sterical surface protection which slows down the macromolecules adsorption to the liposomal surface and hence starch suppresses clearly the elimination of the drug-carrying system from blood by the phagocytes.
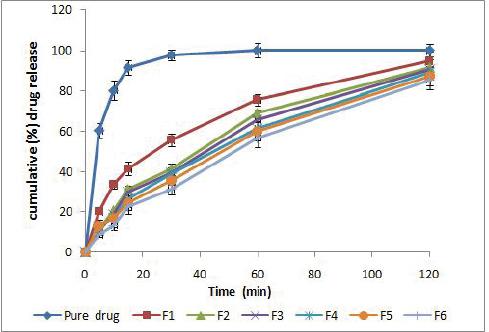
Figure 2 in vitro drug release curve representing cumulative drug release for the pure drug formula and the other sex prepared formula (F1 to F6)
The stability of liposomes at two different storage temperature ranges, 4-8 C, and 20-25 C, was evaluated in terms of size, zeta potential, and entrapment efficiency. As shown in Table 5, F6 stored at 4-8 C did not show any significant change in the amount of entrapped drug; but around 11.5 % drug was lost when stored at 20-25 C for one month while F1 formula after storage at 4-8 C showed a negligible change in the amount of entrapped drug while after storage at 20-25 C about 21.5% drug was lost. Thus at a higher temperature, ALDS was leaching out of the liposomes, that is consistent with earlier observations that at a higher temperature, the permeability and the fluidity of phospholipid bilayer increases27. Earlier reports showed that coating with natural polymers confers stability and enhances the dispersability of particulate systems in water. This may explain the stability of the F6 formula compared with F1.
Table 5 Particle size, zeta potential, and entrapment efficiencies for F1 and F6 liposomal formulas
| Liposomal formula | F1 | F6 | ||||
|---|---|---|---|---|---|---|
| Stage | initial | After one month | Initial | After one month | ||
| Temperature | 4-8 °C | 20-25°C | 4-8 °C | 20-25 °C | ||
| Particle size (nm) | 94-170 | 98-178 | 103-184 | 110-1906 | 134-1993 | 154-2057 |
| Zeta potential (mV) | -12±0.49 | -10±0.89 | -8±0.25 | -39±2.19 | -33±.67 | -27±1.28 |
| Entrapment efficiency (%) | 46.5±1.89 | 43±1.19 | 25±2.16 | 78.5±1.32 | 74.5±1.42 | 67±0.51 |
After one month storage of both F1 and F6, there was a slight increase in the particle size of F6 while there was no significant change in the particle size of F1, confirming the colloidal stability of F1 and F6 formulations. The slight change in the particle size of F6 may be attributed to the slight agglomeration of liposomes. While the zeta potential of F1 didn't show significant change, the zeta potential of F6 showed slight decrease that might be attributed to the coated layer dissociation.
TEM
Liposome morphology was visualized by TEM. Figure 3 shows a liposome outer shape images of the optimum formula (F6) under different magnification power. Cumulative data obtained by TEM showed that the vesicles are well-defined and spherical in shape. There is an agglomeration of excess amounts of starch as appeared in the images, the excess amount of starch was incorporated randomly in the voids between liposomes and results in few agglomerations, that result usually occurred upon using of natural polymers such as starch and chitosan during liposomes preparation28.

Ulcerogenicity study
The results showed that Formula 6 has superior safety and gastric tolerability compared to the pure drug as shown in table 6 and fig. 4. Clearly, Formula 6 demonstrated a remarkable enhancement in ulcer index (UI = 3.05) in comparing with an Alendronate solution (UI = 13.21). Encapsulation of several orally taken active ingredients in liposomes provides protection against both intestinal and gastric ulceration29. Our results were in agreement with Soehngen et al., who proved that the liposomal encapsulation of indomethacin provides an obvious protection against ulceration when it was orally administered to rats30 and also other studies have illustrated that liposomal encapsulation may have a clear effect on reducing GIT side effects e.g ulceration31. Furthermore, starch has an effect for stomach protection, acts as a demulcent and also has a soothing effect on the GIT mucosa32.
Table 6 Ulcerogenic effects of F1 and F6
| Formula | Number of ulcers (average) | Ulcer index* |
|---|---|---|
| Pure drug | 6.5 | 13.21 |
| 6 | 0.6 | 3.05 |
| Control | - | Nil |
*

Figure 4 Pictures of rats stomach mucosa treated with F6(c) compared to F1(B) and control(A)The decrease in side effects of the newly prepared formulas due to several factors including:(i) The syrup-like nature of the current formulas, being of viscous mass, may remain in the first portion of the intestine for a longer time and if administered after cooling in a refrigerator, improved palatability is expected. This also reduces the transit time and hence increases the patient compliance(33).(ii) Colloidal starch has a soothing effect on the stomach and hence decrease the potential gastric upset(34)(ii) ALDS is irritating for stomach(35), and its entrapment into the liposome may have an effect on decreasing its potential side effects.
CONCLUSION
In an attempt to develop an oral liposomal delivery system, natural polymer (starch) was used as means of improving liposomal properties. The liposomes of ALDS were prepared using phosphatidylcholine /cholesterol and different amounts of starch. ALDS /starch mixture was entrapped into the vesicles and excess starch coated the vesicles leading to further improvement of encapsulation efficiency and in-vitro stability. Formulation F6 showed high encapsulation efficiency with minimum particle size and drug release over an 8 h. Furthermore, F6 has shown better gastric tolerability in comparison with the pure drug. Consequently, this new formula of liposomes appears to be an effective drug delivery system for alendronate and with better tolerability and supposed to give greater bioavailability. It is predicted that the novel prepared formulas have fewer side effects due to several factors including:
(i) The syrup-like nature of the current formulas, being of viscous mass, may remain in the first portion of the intestine for a longer time and if administered after cooling in a refrigerator, improved palatability is expected. This also reduces the transit time and hence increases the patient compliance33.
(ii) Colloidal starch has a soothing effect on the stomach and hence decrease the potential gastric upset34
(ii) ALDS is irritating for stomach35, and its entrapment into the liposome may have an effect on decreasing its potential side effects.

