










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkPharmacy Practice (Granada)
versión On-line ISSN 1886-3655versión impresa ISSN 1885-642X
Pharmacy Pract (Granada) vol.4 no.4 Redondela oct./dic. 2006
| Original Research |
Lessons from gefitinib-induced interstitial lung disease in Japan: Problems in approval, pharmacovigilance, and regulatory decision-making procedures
Tsutomu NISHIMURA, Harue TADA, Masatsugu NAKAGAWA, Satoshi TERAMUKAI, Shigeyuki MATSUI, Masanori FUKUSHIMA.
| ABSTRACT Objective: The objective of this study was to identify problems in the approval, pharmacovigilance, and post-approval regulatory decision-making procedures involving gefitinib and to propose countermeasures to prevent further drug-induced suffering in Japan in the future. Key words: Gefitinib. Product surveillance, postmarketing. Lung diseases, interstitial. Decision making, organizational. Japan. | RESUMEN Objetivo: Identificar los problemas en el registro, farmacovigilancia y toma de decisiones post-registro relativas al gefitinib y proponer contramedidas para prevenir futuros problemas producidos por medicamentos en Japón. Palabras clave: Gefitinib. Vigilancia de productos, post-comercialización. Enfermedades pulmonares, intersticiales. Toma de decisiones, organizativas. Japón. |
Tsutomu NISHIMURA. Clinical Trial Design and Management, Translational Research Center, Kyoto University Hospital, Kyoto, (Japan).
Harue TADA. Clinical Trial Design and Management, Translational Research Center, Kyoto University Hospital, Kyoto, (Japan).
Masatsugu NAKAGAWA. Clinical Trial Design and Management, Translational Research Center, Kyoto University Hospital, Kyoto, (Japan).
Satoshi TERAMUKAI. Clinical Trial Design and Management, Translational Research Center, Kyoto University Hospital, Kyoto, (Japan).
Shigeyuki MATSUI. Pharmacoepidemiology, Graduate School of Public Health, Kyoto University, Kyoto, (Japan).
Masanori FUKUSHIMA. Clinical Trial Design and Management, Translational Research Center, Kyoto University Hospital, Kyoto, (Japan).
INTRODUCTION
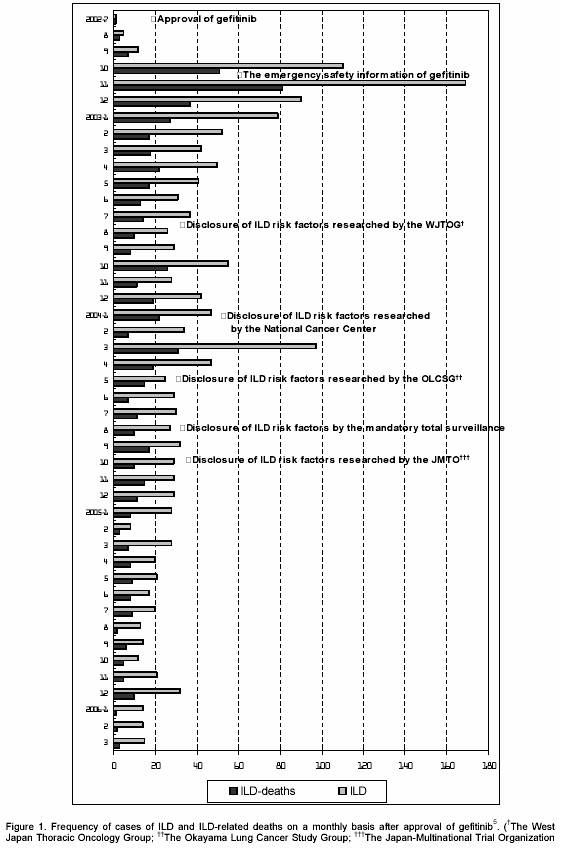
Gefitinib is an oral anticancer molecular-targeted drug that targets epidermal growth factor receptor (EGFR) in tumor cells. As of November 2005, gefitinib had been approved for sale in 36 countries besides Japan.1 In Japan, on July 5, 2002, it was approved for the treatment of advanced non-small cell lung cancer (NSCLC) via an accelerated approval process by the regulatory agency, the Ministry of Health, Labour and Welfare (MHLW), before approval in any other country. The basis for approval was mainly the high response rate (26.5%) found in Japanese phase II trials.2 Within four months of approval, however, 87 deaths due to interstitial lung disease (ILD) including interstitial pneumonia had been disclosed in spontaneous reports in Japan.3 Figure 1 shows the frequency over time of cases of ILD and ILD-related deaths after approval. As of December 2004, approximately 44,000 patients have been treated in Japan since the approval of gefitinib and as of March 2006 the total number of ILD-related deaths reached 643.4,5 The incidence of ILD and ILD death related with gefitinib were identified 5.8% and 2.3%, respectively by the mandatory total registry and surveillance of all drug users (mandatory total surveillance) which was conducted by AstraZeneca from June 2003 to March 2004 in Japan.6 The data besides Japan was summarized as follows by Evans.7 Of the 44,000 patients treated outside Japan, 0.33% developed pneumonitis and 0.1% developed fatal pneumonitis. As part of the expanded access program (EAP) in the US, 24,000 patients received gefitinib prior to its FDA approval. Pneumonitis occurred in 0.36% of patients and fatal pneumonitis occurred in 0.06%. Similar percentages were noted in the EAP conducted throughout the rest of the world (14,500 patients), with 0.35% of patients developing pneumonitis and 0.06% developing fatal pneumonitis. Globally, there have been at least 80,000 patients treated with gefitinib; 1% of these patients developed interstitial pneumonitis and 0.37% developed fatal interstitial pneumonitis. Autopsy data from cases of fatal pneumonitis are limited (approximately 3% of cases). In those cases examined, the lungs were characterized by diffuse alveolar damage. In Japan, the incidences of 5.8 % ILD and 2.3 % ILD death related with gefitinib had become a serious social issue. The gefitinib label was revised seven times in the 18 months after drug approval,8 but major revisions only related to data reported before approval. In July 2004, the family of a patient who had died from ILD allegedly associated with gefitinib filed a lawsuit against the government and AstraZeneca, the manufacturer of gefitinib, claiming that the patient had suffered from adverse drug reactions (ADR). In spite of the label being revised numerous times, as of March 2006, approximately 10 ILD death related with gefitinib were still being reported each month in Japan.5

In the present article, we critically review the approval, pharmacovigilance, and regulatory decision-making procedures for gefitinib in Japan, and identify problems therein. We then propose countermeasures to prevent further serious drug-induced suffering in Japan.
METHODS
We systematically reviewed gefitinib-related reports published between July 2000 and March 2006 by Japanese and the US regulatory agencies (i.e. the Japanese MHLW,4,5,8-11 the Japanese Pharmaceuticals and Medical Devices Evaluation Center,12 and the US Food and Drug Administration [FDA]13), AstraZeneca,1-3,6,14-25 cancer clinical study groups,26-30 and by a scientific society (the Japan Lung Cancer Society).31,32 In particular, we focused on documents regarding New Drug Applications (NDA) for approval, including the results of animal experiments and clinical trials before approval,33-36 the results of the EAP, and data regarding adverse events (AE) reported in Japan and other countries. On the basis of information in the gefitinib label in Japan14 and the US,19 the Physicians Desk Reference (PDR)37 and the FDA Drug Approval Summary,13 ILD may have been used to define the type of pulmonary toxicity found with gefitinib. ILD comprises a large number of conditions that involve the parenchyma of the lung, including the alveoli, the alveolar epithelium, the capillary endothelium, and the spaces between these structures, as well as the perivascular and lymphatic tissues.38 ILD is difficult to diagnose.39-41 The diagnosis of drug-associated lung disease is against the background of the underlying neoplastic lung disease, adverse effects of other drugs, colony-stimulating factors, oxygen or radiation therapy and opportunistic infections.40 Clinical symptoms of ILD, such as escalating dyspnea, cough, and fever, may be indistinguishable from the symptoms of progressive tumor growth or infection. Computed tomography features of ILD include pulmonary reticular changes and ground-glass opacity, which are also nonspecific and may not readily indicate a precise etiology.39 Diagnosis of drug-induced ILD thus relies on rigorous exclusion of all other differential diagnoses.39,42,43 During the post-marketing surveillance of gefitinib, AstraZeneca defined ILD as comprising interstitial pneumonia, bronchopneumonia, pneumonia, pneumothorax, acute respiratory distress syndrome, acute respiratory failure, acute eosinophilic pneumonia, dyspnea, breathing disorder, respiratory failure, hypoxia, pulmonary hemorrhage, pulmonary disorder, pulmonary infiltrates, pulmonary fibrosis, pneumonitis, alveolus hemorrhage, PO2 reduction, oxygen saturation reduction, or radiation pneumonitis.21 It was probably used to make certain all possible cases were included. The mandatory total surveillance was conducted by AstraZeneca from June 2003 to March 2004.6 The mandatory total surveillance is a single cohort study that all targeted drug users are registered and are followed up for planed period and all or targeted AE are researched. Because ILD is difficult to diagnose, diagnosis of drug-induced ILD relies on rigorous exclusion of all other differential diagnoses. In mandatory total surveillance, all patients were diagnosed based on the strict diagnostic procedures to exclude all other differential diagnoses.6 In the first stage of the procedures, radio-diagnosis specialists diagnosed whether lung diseases were ILD or interstitial pneumonia, absence of ILD or interstitial pneumonia, or unable to evaluate based on image information. In the second stage, the cases diagnosed as absence of ILD or interstitial pneumonia or unable to evaluate in the first stage were diagnosed by two respiratory physicians whether lung diseases were ILD or interstitial pneumonia, absence of ILD or interstitial pneumonia, or unable to evaluate based on image and clinical information. In the third stage, the cases diagnosed as absence of ILD or interstitial pneumonia or unable to evaluate in the second stage were diagnosed by the assessment committee whether lung diseases were ILD or interstitial pneumonia, absence of ILD or interstitial pneumonia, or unable to evaluate based on imaging and clinical criteria (Table 1).6 As a result, the incidence of ILD and ILD death related with gefitinib were identified 5.8% and 2.3%, respectively.6
RESULTS
Detailed information of toxicity studies with animals, conducted by AstraZeneca before approval, was released on February 25, 2005.23 According to that information, in a six-month oral toxicity study in dogs, the gefitinib administration groups showed three instances of alveolar macrophages, one chronic pneumonia, one focal alveolar septal metaplasia, and one focal alveolar haemorrhage.23 Although those findings were not observed in the placebo group,23 the information was not reflected in the new drug application summary nor in the first edition of the label.2,14 Other experiments showed that gefitinib aggravated pulmonary fibrosis in a bleomycin-induced murine model.35 Although these findings were reported to AstraZeneca before May 2002, AstraZeneca did not report that information to MHLW.17,36 From clinical trials conducted in Japan and other countries, 90 pulmonary serious adverse events (SAE) and 17 pulmonary SAE-related deaths in total were reported before approval of the drug (Table 2).2 In the same trials, seven of the 90 pulmonary SAE were identified as ADR, and the other cases were found to be AE unrelated to the drug.2 Although pulmonary serious AE do not necessarily equal ILD, in the pre-marketing clinical trials, ILD were not distinguished from pulmonary serious AE. In Japan, 133 patients were administered gefitinib in phase I, II, and those continuous administration clinical trials.2 In those trials, three cases of interstitial pneumonia and four cases of pneumonia were reported as AE.2,12 Among the AE, one case of interstitial pneumonia and one case of pneumonia were ADR.2 However, on May 9, 2002 the Pharmaceuticals and Medical Devices Evaluation Center researched 3 interstitial pneumonia cases and stated that it was undeniable that gefitinib was related to development of ILD.12 Therefore, the three cases of interstitial pneumonia were ADR. According to data from phase III clinical trials and the EAP conducted in other counties, 40 out of the reported 196 cases were pulmonary ADR.10 In addition, 22 out of 56 ADR deaths were pulmonary ADR.10 Thereby, the greatest cause of deaths was pulmonary ADR.10 Although pulmonary ADR are clinically very serious problems, the first edition of the label did not contain a caution relating to the potential for pulmonary ADR including interstitial pneumonia.14 Although at least one pneumonia was ADR related with gefitinb,2 for pulmonary ADR, only interstitial pneumonia was noted as being an important ADR in the label when the drug was released.14 In addition, the label claimed that the frequency of interstitial pneumonia was unknown,14 whereas it could be calculated as being 2.3% (3/133) from the data reported before approval. Thus, the safety data regarding lungs were not properly reflected in the form of warnings in the first edition of the label.14

On July 5, 2002, when it was first approved in Japan, gefitinib was approved for the treatment of inoperable or recurrent NSCLC,14 whereas when the drug was approved in the US on May 5, 2003, it was approved for the treatment of locally advanced or metastatic NSCLC that responded to neither platinum-based nor docetaxel chemotherapy.19 The inclusion criteria in phase II clinical trials were (1) presence of advanced NSCLC and previous treatment with one or two chemotherapy regimens (containing at least one platinum-based agent) (IDEAL1 in Japan, Europe, Australia, and South Africa)33 and (2) presence of recurrent NSCLC and previous treatment with two or more chemotherapy regimens containing a platinum-based agent and docetaxel, given concurrently or sequentially (IDEAL2 in the US).34 However, in Japan the MHLW expanded the indications beyond the eligibility criteria of the clinical trials.
Table 3 lists the post-marketing surveillance conducted in Japan on gefitinib. According to emergency safety information released by the MHLW on October 15, 2002, as of October 11, 2002, in Japan 22 ILD and 11 ILD-related deaths in total had been reported.15 In fact, as of October 5, 2002, a total of 49 ILD and 24 ILD-related deaths had been reported in spontaneous reports.3 On October 29, 2002, one of the authors of the present article (Fukushima M) pointed out that all ADR cases should be disclosed and analyzed to identify risk factors and mandatory total surveillance should be immediately performed.44 However, that opinion was ignored. In January 2003, the first report regarding gefitinib-induced ILD in Japan was published.26 On February 28, 2003, Hama, Beppu, and Fukushima requested MHLW to withdraw approval of gefitinib and recall the drug from the Japanese market because risks of gefitinib were thought to be greater than the benefits.45 But that suggestion was not accepted by the MHLW at all. In the US, on May 1, 2003, the American Public Citizens Health Research Group in a letter to the FDA expresses concern at the pending approval of gefitinib because the pivotal trial on which accelerated approval for gefitinib rests is a small uncontrolled, unblinded, Phase II trial in atypical NSCLC patients for third-line treatment and a well-conducted Phase III trial of gefitinib as first-line therapy in NSCLC patients was unequivocally negative with respect to all endpoints.46 On May 5, 2003, however, FDA approved gefitinib. Meanwhile, the incidence rate of ILD was not actually estimated at that time because AstraZeneca did not report the exact number of patients that had used gefitinib. On January 20, 2005, AstraZeneca estimated that the cumulative total of gefitinib users in Japan was 86,800,4 but on March 24, 2005, this estimate was halved to 42,000,24 with the result that the incidence rate of ILD doubled.

On December 5, 2002, AstraZeneca convened a meeting to discuss instances in which patients who received gefitinib treatment had developed ILD. On March 26, 2003, AstraZeneca released a list of factors that related to higher risk of deaths for ILD developed patients. The list included male sex, the presence of squamous cell carcinoma, and history of idiopathic pulmonary fibrosis.18 However, risk factors for ILD development were studied in the meeting convened by AstraZeneca. In June 2003, mandatory total surveillance in Japan started about one year after approval of the drug.6
On July 18, 2003, the West Japan Thoracic Oncology Group reported risk factors for ILD, including male sex, a history of smoking, and existence of idiopathic pulmonary fibrosis.27 On October 20, 2003, the Japan Cancer Society released the Statement on gefitinib suggesting that according to the results of studies by AstraZeneca and the West Japan Thoracic Oncology Group, the use of gefitinib for higher risk patients should be limited to patients in whom the benefits were greater than the risks.31 In January 2004, investigators at the National Cancer Center in Japan reported that pre-existing pulmonary fibrosis was a risk factor for ILD.28 In June 2004, the Okayama Lung Cancer Study Group reported risk factors for ILD, including presence of pulmonary fibrosis before gefitinib treatment and poor performance status.29 In August 2004, as a result of the mandatory total surveillance, risk factors for ILD, including the presence of performance status 2-4, a history of smoking, previous co-existence of ILD, and history of chemotherapy were identified about two years after approval.6 In October 2004, the Japan-Multinational Trial Organization reported risk factors for ILD, including a decrease in serum albumin and absence of a history of chemotherapy.30
DISCUSSION
In Japan, gefitinib was approved on July 5, 2002, for the treatment of NSCLC, through an accelerated approval process by the Japanese regulatory agency (the MHLW), before approval in any other country. However, many deaths from ILD related with the use of gefitinib have been reported. The high incidence of and mortality from ILD became a serious social issue in Japan. We identified three major problems in the approval, pharmacovigilance, and regulatory decision-making procedures for gefitinib in Japan, through a critical review of reports regarding gefitinib published by regulatory agencies, AstraZeneca, cancer clinical study groups, and a scientific society during the period from 2000 to 2006. The problems we identified are: 1) the results of animal experiments, pre-marketing clinical trials, and reports of ADR from other countries were not properly reflected in the label; 2) indications for use of the drug were expanded without strict evaluation of the external validity of pre-marketing clinical trials; and 3) despite many cases of serious ILD being spontaneously reported, well-designed post-marketing surveillance was not immediately performed.
With respect to the first problem, all serious safety events reported before approval, from animal experiments, pre-marketing clinical trials, and reports of ADR from other countries and so on, should be properly reflected in the label, irrespective of whether or not causality has been attributed to the drug. At the same time, prompt and thoroughgoing distribution of important safety information to the public must be ensured by several effective means, including official announcement of emergency safety information by the MHLW. While in Japan gefitinib induced problems as described above, outside Japan, rofecoxib that was Merck and Co's leading drug for control of acute pain, and pain associated with osteoarthritis, rheumatoid arthritis, and menstruation induced problems. On September 30, 2004, Merck withdrew rofecoxib after the trial, APPROVe (Adenomatous Polyp Prevention On Vioxx), showed an cardiovascular ADR profile.47 Merck was indeed fully aware of rofecoxib's potential risks by 2000.48 For that problems, Dieppe et al. suggested that legal requirement for drug companies to make all data on serious adverse events from clinical studies publicly available immediately after study completion.49 This suggestion corresponds with our opinion.
Regarding the second problem, indications must be determined in an appropriate manner at approval through strict evaluation of the external validity of clinical trials. Any expansion of the indications beyond the eligibility criteria of pre-marketing clinical trials must be accompanied by careful post-marketing administration and rigorous monitoring for patients that did not meet the original criteria. On October 20, 2003, the Japan Lung Cancer Society released a Statement on Gefitinib, which suggested that the inclusion and exclusion criteria in IDEAL1 should be taken as the as the selection criteria for patients to be treated with gefitinib.31 Furthermore, it was suggested that gefitinib should not be used for the treatment of other patients, except in clinical trial setting, because the safety of the gefitinib treatment had not been evaluated for these patients.31 This stance ought to have been officially adopted with respect to the indications at approval.
Regarding the third problem, when unexpected serious AE are reported after approval of a drug, well-designed post-marketing surveillances should be performed immediately to collect precise information about the incidence of AE and the associated risk factors. In International Conference on Harmonisation (IGH) E2E, at the beginning of the Pharmacovigilance plan a summary should be provided of the: important identified risks, important potential risks, and important missing information.50 In the gefitinib case, 3 interstitial pneumonia reported before the approval were thought to be important identified risks. Despite many cases of serious ILD including interstitial pneumonia being spontaneously reported, they were not regarded as a signal. In the result, the mandatory total surveillance was not immediately performed. When the emergency safety information released by the MHLW on October 15, 2002, ILD should have been regarded as a signal and the mandatory total surveillance should have been performed. Some approaches that use various existing databases have been proposed in the pharmacoepidemiological literature for post-marketing surveillance,51,52 including the combination of hospital-based databases and population-based databases. However, these approaches can have serious limitations with respect to bias (selection and information bias) because these existing databases were originally established for purposes other than collecting precise information about the incidence of safety events and the risk factors for a specific drug of interest. An alternative approach to overcome these limitations is a prospective study of all drug users. For estimating the incidence rate, well-designed mandatory total surveillance would allow determination of the incidence rate exactly, without invoking complex estimation procedures with unverifiable assumptions for selection and information biases. On the contrary, although a week point of mandatory total surveillance is to need high cost, use of information technology will lead to low cost mandatory total surveillance.
In Japan, the regulatory agency, the MHLW can enforce mandatory total surveillance based on Pharmaceutical Affairs Law, which has been adopted for approved drugs for which the number of Japanese users is small and/or the incidence rate of serious ADR (e.g. bone-marrow toxicity) is expected to be high.11 The anticancer drug irinotecan, a camptotecin analogue manufactured by the Yakult Company, and which was approved in Japan on January 19, 1994, was the first drug for which the mandatory total surveillance system was adopted. The main toxic effects of irinotecan were grade 3 or 4 diarrhea and leukopenia. For 414 patients in the phase I and II trials, the incidence rates of diarrhea and leukopenia of grade 3 or 4 according to the Japan Society of Clinical Oncology Criteria53 were 17.1% and 32.4%, respectively (Table 4).54 Such a high incidence rate of these serious events was one of the main reasons for adopting the mandatory total surveillance system. Another drug for which this system was adopted was TS-1, an anticancer drug manufactured by the Taiho Pharmaceutical Company that was approved on January 25, 1999. Unlike conventional oral fluorouracil drugs, the dose-limiting toxicity of TS-1 was bone marrow depression. For 578 patients in the phase I and II trials, the incidence rates of leukopenia, neutropenia, and thrombocytopenia of grade 3 or 4 according to the Japan Society of Clinical Oncology Criteria53 were 2.8%, 8.5%, and 1.6%, respectively (Table 4).55 For gefitinib, the incidence rate of interstitial pneumonia for 133 patients in the phase I, II, and those continuous administration clinical trials was 2.3%, which is comparable to the rates of serious ADR related with TS-1 (Table 4). Furthermore, the number of patients used for safety assessment was 133 for gefitinib, much smaller than the 414 used for irinotecan and the 578 used for TS-1. Although the MHLW argued that adoption of mandatory total surveillance was not necessary for gefitinib because the conditions for invoking mandatory total surveillance in terms of the number of patients treated and the incidence rates of serious ADR (interstitial pneumonia) had not been met,11 obviously this argument is not supported by the aforementioned facts from phase I and II trials. When other anti-tumor agents are administered intravenously, blood tests are performed before administration to exclude patients in whom the therapy would be inappropriate. In addition, those agents are periodically administered and the blood level of agents may be minimum when ADR develop. However, because gefitinib is an oral drug and is taken daily without a blood test, the blood level of gefitinib may peak and remain high when ILD develops. Because of the route of administration of gefitinib, this is a particular risk. Moreover, many irregular usages are expected when an oral drug is used in real-world situations for advanced NSCLC patients, for whom effective treatments have not been well established. Hence, it is our opinion that mandatory total surveillance ought to have been planned at approval for gefitinib.

Because of the limited number of patients in pre-marketing trials and substantial gaps between the pre-marketing ideal protocol environment and post-marketing real-world situations, a well-established system of post-marketing pharmacovigilance and surveillance is absolutely indispensable to prevent serious drug-induced suffering. We propose establishment of mandatory total registry of all drug users and surveillance as one of the rational solutions for preventing further drug-induced suffering in Japan. The sample size for mandatory total surveillance should be carefully determined depending on the drug, the targeted disease, and the purpose of surveillance. All users must be registered and be followed up until a sufficient sample size is reached, and then outcomes should be surveyed to identify risk factors for SAE.
Cohort or case-control studies of both exposed and unexposed patients for a particular treatment are useful for evaluating causal relationship of AE to the treatment, and possibly to other factors. A nested-case control study to determine the relative risk of and risk factors for ILD in a cohort of NSCLC patients treated with and without gefitinib is ongoing in Japan.56 Under possible interaction between treatment and risk factors that the effect of risk factors on AE is modified by treatment, the mandatory total surveillance would be more relevant to identify a high risk group who needs meticulously careful administration and thorough monitoring, particularly, in patients for whom the treatment is limited.
As a new molecular-targeted agent, gefitinib is of interest to physicians and patients because it could possibly effect disease stabilization and symptom control in some symptomatic patients, but although it has been approved in many countries, the survival benefits of this drug are still under investigation. The results of a multinational placebo-controlled trial, the Iressa Survival Evaluation in Lung cancer (ISEL) study, were released on December 17, 2004.57 Because a survival benefit attributable to gefitinib relative to a placebo was not demonstrated in that study, AstraZeneca restricted the use of gefitinib in the US, and withdrew its new drug application for approval in Europe.58,59 More recently, a randomized clinical trial (S0023) that was sponsored by the National Cancer Institute and conducted by the Southwest Oncology Group (Ann Arbor, Michigan) and AstraZeneca was closed after interim analysis because no survival benefit was found compared with a placebo following chemotherapy and radiation for patients with NSCLC that had spread only to nearby tissues or lymph nodes.60 In addition, the Second Line Indication of Gefitinib in NSCLC (SIGN) study, which compared gefitinib with docetaxel as second-line therapy for advanced NSCLC, was also conducted. The results of that study suggested that gefitinib is likely to be comparable to docetaxel in terms of median survival, but is better tolerated.61 In Japan, official meetings to review the results of the ISEL study were convened from December 2004 to March 2005. In the ISEL study, subgroup analyses suggested some survival benefit in patients of oriental origin.57 In addition, more EGFR mutations were detected in advanced NSCLC patients who were female, had adenocarcinoma and were non-smokers, and Japanese women had a high response rate.62-64 A retrospective study showed that patients with EGFR mutations survived for a longer period than those without the mutations after initiation of gefitinib treatment.65 Based on these results, the Japan Lung Cancer Society provided new guidelines on February 19, 2005,32 recommending that gefitinib be used for the treatment of adenocarcinoma, women, non-smokers, Japanese, or patients with an EGFR mutation. The regulatory agency (the MHLW) adopted these guidelines for appropriate use of the drug and decided not to withdraw the drug from the Japanese market.9 One flaw in the process of the official meeting to discuss the results of the ISEL study was that the person in charge, the chief of the Safety Division of the MHLWs Pharmaceutical and Food Safety Bureau,66 was also responsible for approval of gefitinib as the Chief of the Office of New Drugs I in the Pharmaceuticals and Medical Devices Evaluation Center when the drug was under review as a new drug application in May 2002.67 Such defect in the Japanese system for development, approval and dispensing of drugs has not been rectified since one of the authors of the present article first pointed out this problem 17 years ago, as something Japan has not learned from the lessons of past drug disasters.68,69 This system must be rectified.
While Japan struggled with ongoing reports of gefitinib-induced ILD deaths without definitive evidence of a survival benefit of the drug, AstraZeneca restricted the use of gefitinib in the US and withdrew their application for approval in Europe because of a lack evidence of survival benefit.70 An AstraZeneca-sponsored phase III study comparing gefitinib with docetaxel as second or third-line therapy is ongoing in Japan.71 But to obtain definitive conclusions with respect to effectiveness and safety, a randomized trial is required to compare gefitinib with placebo treatment for patients who are refractory to standard therapy. On the other hand, ILD has also been reported as an ADR for erlotinib, which is the only EGFR-inhibitor available in the US and European markets for which there is evidence of a survival benefit in lung cancer.72,73 It was very recently disclosed in a pancreatic cancer study that in combination with gemcitabine, the incidence of ILD-like events was 2.5% in the erlotinib plus gemcitabine group, versus 0.4% in the placebo plus gemcitabine group.73 Moreover, in a phase I study involving 15 Japanese patients, one patient had grade 5 interstitial pneumonia.74 In addition, a case report showed that ILD was induced by erlotinib in a patient who had previously tolerated gefitinib.75 These facts suggest that ILD is common to all EGFR-inhibitors. Erlotinib has been approved in the US and Europe, but it is not approved in Japan. Because there is evidence that erlotinib confers a survival benefit, the authorities have been urged to approve erlotinib in Japan. It is no question that ILD caused by the drug should be carefully monitored; for example, mandatory total surveillance ought to planned after approval of erlotinib. In addition, after approval of erlotinib, a randomized trial to compare gefitinib with erlotinib as second or third-line therapy is required in order to determine which EGFR-inhibitor is safer and more effective for Japanese patients.
DISCLAIMER
This study was conducted without any external funding.
| References |
1. AstraZeneca. Selling countries abroad until November 2005. (in Japanese). http://www.iressa.jp/professional/faq/pro_faq1.asp#01_02 (accessed 2006 August 2) [ Links ]
2. AstraZeneca. Information about Iressa tablets 250. (in Japanese). http://www.info.pmda.go.jp/shinyaku/g020706/index.html? (accessed 2006 August 2) [ Links ]
3. AstraZeneca. The trend in the incidence of interstitial lung diseases and interstitial pneumonia. February 10, 2003 (in Japanese). http://www.astrazeneca.co.jp/activity/other/detail/03_02_10b.html (accessed 2006 August 2) [ Links ]
4. Ministry of Health, Labour and Welfare. The trend in the incidence of interstitial lung diseases and interstitial pneumonia possibly related to the use of gefitinib reported to Ministry of Health, Labour and Welfare and Pharmaceuticals and Medical Devices Agency: Tabulation by the day of reporting until December 28, 2005. January 20, 2005 (in Japanese). http://www.mhlw.go.jp/shingi/2005/01/dl/s0120-4b.pdf (accessed 2006 August 2) [ Links ]
5. Ministry of Health, Labour and Welfare. The reported number of interstitial lung diseases and interstitial pneumonia after gefitinib therapy. April 26, 2006 (in Japanese). http://www.mhlw.go.jp/houdou/2006/04/h0426-1.html (accessed 2006 March 30) [ Links ]
6. AstraZeneca. The results and discussions of the prospective study (Special Surveillance) for Iressa® tablets 250. August 2004 (in Japanese). http://www.mhlw.go.jp/shingi/2005/01/s0120-4.html (accessed 2006 March 30) [ Links ]
7. Evans TL. Highlights from the Tenth World Conference on Lung Cancer: The Oncologist 2004; 9 (2), 232–238. [ Links ]
8. Ministry of Health, Labour and Welfare. State of the implementation of measures for the safe use of Iressa, including disclosure of safety information. January 20, 2005 (in Japanese). http://www.mhlw.go.jp/shingi/2005/01/dl/s0120-4d.pdf (accessed 2006 August 2) [ Links ]
9. Ministry of Health, Labour and Welfare. An opinion on evaluation of the results of ISEL study and future countermeasures for the use of Iressa. March 24, 2005 (in Japanese). http://www.mhlw.go.jp/shingi/2005/03/s0324-12.html (accessed 2006 August 2) [ Links ]
10. Ministry of Health, Labour and Welfare. A list of adverse drug reactions reported before approval of Iressa. December 25, 2002 (in Japanese). http://www.mhlw.go.jp/shingi/2003/05/dl/s0502-1m1.pdf (accessed 2006 August 2); http://www.mhlw.go.jp/shingi/2003/05/dl/s0502-1m2.pdf (accessed 2006 August 2) [ Links ]
11. Ministry of Health, Labour and Welfare. Minutes of the fourth official meeting to discuss the gefitinib problem. March 24, 2005 (in Japanese). http://www.mhlw.go.jp/shingi/2005/03/txt/s0324-6.txt (accessed 2006 August 2) [ Links ]
12. Pharmaceuticals and Medical Devices Evaluation Center. The report of new drug application review for Iressa. May 9, 2002 (in Japanese). http://211.132.8.246/shinyaku/g0207/06/67022700_21400AMY00188_110_1.pdf (accessed 2006 August 2) [ Links ]
13. FDA Drug Approval Summary: Gefitinib (ZD1839) (Iressa®) Tablets. Oncologist 2003; 8: 303-306. [ Links ]
14. AstraZeneca. Label for Iressa® 250 tablets, first edition. July 2002 (in Japanese). [ Links ]
15. AstraZeneca. The Emergency Safety Information for Iressa. October 15, 2002 (in Japanese). http://www.info.pmda.go.jp/kinkyu_anzen/kinkyu20021015.pdf (accessed 2006 August 2) [ Links ]
16. AstraZeneca. A report of early post-marketing pharmacovigilance: Iressa® tablets 250. March 2003 (in Japanese). [ Links ]
17. AstraZeneca. Comments on recent news reports. March 12, 2003 (in Japanese). http://www.astrazeneca.co.jp/activity/other/detail/03_03_11.html (accessed 2006 August 2) [ Links ]
18. AstraZeneca. The final report of the Professional Meeting regarding interstitial lung diseases and interstitial pneumonia induced by the use of gefitinib (Iressa® tablets). March 26, 2003 (in Japanese). [ Links ]
19. AstraZeneca. Label for IressaTM (gefitinib tablets) 250mg, first edition. May 2, 2003. http://www.fda.gov/cder/foi/label/2003/021399lbl.pdf (accessed 2006 August 2) [ Links ]
20. AstraZeneca. A report of monitoring of adverse drug reaction: Iressa® tablets 250. September 2003 (in Japanese). [ Links ]
21. AstraZeneca. A report of adverse drug reaction: Iressa® tablets 250. October 2003 (in Japanese). [ Links ]
22. AstraZeneca. A list of adverse drug reactions: Iressa® tablets 250. April 2004 (in Japanese). [ Links ]
23. AstraZeneca. The report of general toxicity studies. February 25, 2005. http://med.astrazeneca.co.jp/iressasp/index.html (accessed 2006 August 2) [ Links ]
24. AstraZeneca. The estimated number of patients administered Iressa tablets. March 24, 2005 (in Japanese). http://www.mhlw.go.jp/shingi/2005/03/dl/s0324-11d.pdf (accessed 2006 August 2) [ Links ]
25. AstraZeneca. The answer about EGFR mutation related iressa from AstraZeneca. March 17, 2005 (in Japanese). http://www.mhlw.go.jp/shingi/2005/03/s0317-4.html (accessed 2006 August 2) [ Links ]
26. Inoue A, Saijo Y, Maemondo M, et al. Severe acute interstitial pneumonia and gefitinib. Lancet 2003; 261: 137-9. [ Links ]
27. Seto T, Yamamoto N. Interstitial lung diseases (ILD) induced by gefitinib in patients with advanced non-small cell lung cancer (NSCLC): Results of a West Japan Thoracic Oncology Group (WJTOG) epidemiological survey. proc ASCO 2004; (Abstract#7064). [ Links ]
28. Takano T, Ohe Y, Kusumoto M, et al. Risk factors for interstitial lung disease and predictive factors for tumor response in patients with advanced non-small cell lung caner treated with gefitinib. Lung Cancer 2004; 45: 93-104. [ Links ]
29. Hotta K, Harita S, Bessho A, et al. Interstitial lung disease (ILD) during gefitinib treatment in Japanese patients with non-small cell lung cancer (NSCLC): Okayama Lung Cancer Study Group. proc ASCO 2004; (Abstract#7063). [ Links ]
30. Nakagawa M, Teramukai S, Tada H, et al. Hypoalbumia as a risk factor of interstitinal lung disease (ILD) during gefitinib treatment in patients with non-small cell lung cancer (NSCLC) : A JMTO study. proc ASCO 2005; 23: 667s. (Abstract#7190). [ Links ]
31. Japan Lung Cancer Society. Statement on gefitinib. Japanese Journal of Lung Cancer 2003; 43: 780-4 (in Japanese). [ Links ]
32. Japan Lung Cancer Society. The guideline for Iressa use. February 19, 2005. (in Japanese). [ Links ]
33. Fukuoka M, Yano S, Giaccone G, et al. Multi-Institutional Randomized Phase II Trial of Gefitinib for Previously Treated patients With Advanced Non-Small-Cell Lung Cancer (The IDEAL 1 Trial), J Clin Oncol 2003; 21: 2237-2246. [ Links ]
34. Natale RB, Skarin A, Maddox AM, et al. Improvement in symptoms and quality of life for advanced non-small-cell lung cancer patients receiving ZD1839 in IDEAL 2, Proc ASCO 2002; 21:1167. [ Links ]
35. Suzuki H, Aoshiba K, Yokohori N, Nagai A. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibition Augments a murine Model of Pulmonary Fibrosis. Cancer Res 2003; 63: 5054-5059. [ Links ]
36. Mainichi Newspapers. Concealment of the results of animal experiments. February 7, 2003 (in Japanese). [ Links ]
37. Physicians Desk Reference® (PDR®), 60 edition. Thomson, 2006; 685-686. [ Links ]
38. Kasper D, Fauci A, Longo D, Braunwald E, Hauser S, Jameson L. Harrison's Principles of Internal Medicine, 16th edition. McGraw-Hill, 2005; 1554. [ Links ]
39. Danson S, Blackhall F, Hulse P, Ranson M. Interstitial lung disease in lung cancer - Separating disease progression from treatment effects. Drug Saf 2005;28(2):103-13. [ Links ]
40. Camus P. Interstitial lung disease in patients with non-small-cell lung cancer: causes, mechanisms and management. Br J Cancer. 2004 Aug;91 Suppl 2:S1-2. [ Links ]
41. Camus P, Fanton A, Bonniaud P, Camus C, Foucher P. Interstitial lung disease induced by drugs and radiation. Respiration. 2004;71(4):301-26. [ Links ]
42. Ando M, Okamoto I, Yamamoto N, et al. Predictive factors for interstitial lung disease, antitumor response, and survival in non-small-cell lung cancer patients treated with gefitinib. J Clin Oncol. 2006 Jun 1;24(16):2549-56. [ Links ]
43. Camus P, Kudoh S, Ebina M. Interstitial lung disease associated with drug therapy. British Journal of Cancer, (2004), 91, (Suppl 2). [ Links ]
44. Inside Nikkei Biobiz 2002/10/29. Iressa and interstitial pneumonia. (in Japanese). [ Links ]
45. Hama Rokuro, Beppu Hirokuni, Fukushima Masanori. The request for cancel of gefitinib. February 28, 2003 (in Japanese). http://www.npojip.org/iressa/iressa10-r1.html (accessed 2006 August 2) [ Links ]
46. Janet Woodcock. Letter to FDA expressing concerns about the pending approval of the cancer drug gefitinib (IRESSA) (HRG Publication #1665). May 1, 2003. http://www.citizen.org/publications/print_release.cfm?ID=7242 (accessed August 2, 2006) [ Links ]
47. Merck & Co., Inc. Merck & Co., Inc. is a global research-driven pharmaceutical company dedicated to putting patients first. September 30, 2004. http://www.vioxx.com/rofecoxib/vioxx/consumer/index.jsp (accessed August 2, 2006) [ Links ]
48. Horton R. Vioxx, the implosion of Merck, and aftershocks at the FDA. Lancet. 2004; 364: 1995-6. [ Links ]
49. Dieppe PA, Ebrahim S, Martin RM, Juni P. Lessons from the withdrawal of rofecoxib. BMJ 2004;329:867-868. [ Links ]
50. ICH harmonized tripartite guideline pharmacovigilance planning E2E. November 18, 2004. http://www.nihs.go.jp/dig/ich/efficacy/e2e/e2e_041118_e.pdf (accessed August 2, 2006)) [ Links ]
51. Strom BL (ed.). Pharmacoepidemiology (3rd edn), John Wiley & Sons Inc, 2000. [ Links ]
52. Hartzema AG, Porta MS, Tilson HH. Pharmacoepidemiology: An Introduction (3rd edn). Harvey Whitney Books, 1998. [ Links ]
53. J. Jpn. Soc. Cancer Ther. 1986; 21(5): 929-942 (in Japanese). [ Links ]
54. New Drug Approval Summary No.1 Irinotecan hydrochloride. 1st. Society of Japanese Pharmacopoeia, 1994; 47-49: (in Japanese). [ Links ]
55. Taiho Pharmaceutical. Drug Interview Form TS-1 capsule 20·25, 10th. 2004; 112 (in Japanese). [ Links ]
56. Fukuoka M, Kudo S, Kato H, Nakada K, Nishiwaki Y. A nested case-control study to determine the relative risk of and risk factors for interstitial lung disease in a cohort of NSCLC patients treated with and without gefitinib. Medicine and Drug Journal 2005; 41, 790-795. (in Japanese). [ Links ]
57. Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005; 366: 1527-37. [ Links ]
58. AstraZeneca. AstraZeneca Announces US Label Change For IRESSA® (gefitinib) And New Distribution Program. June 17, 2005. http://www.astrazeneca-us.com/modules/PRMS/display.asp?id=571650 (accessed 2006 August 2) [ Links ]
59. AstraZeneca. Gefitinib (IressaTM) marketing authorisation application withdrawn in EU. January 4, 2005. http://www.astrazeneca.com/pressrelease/4442.aspx (accessed 2006 August 2) [ Links ]
60. National Cancer Institute. Clinical Trial of Gefitinib for Advanced Lung Cancer Closes Early. April 18, 2005. http://www.nci.nih.gov/newscenter/pressreleases/gefitinibNSCLC (accessed 2006 August 2) [ Links ]
61. Cufer T, Vrdoljak E. Results from a Phase II, open-label, randomized study (SIGN) comparing gefitinib with docetaxel as second-line therapy in patients with advanced (stage IIIb or IV) non-small-cell lung cancer. proc ASCO 2005; 23: 629s. (abstract#7035). [ Links ]
62. Shigematsu H, Lin L, Takahashi T, et al. Clinical and biological features associated with epidermal growth factor receptor gene mutations in lung cancers. J Natl Cancer Inst 2005 ; 97: 339-346. [ Links ]
63. Kosaka T, Yatabe Y, Endoh H, Kuwano H, Takahashi T, and Mitsudomi T. Mutations of the Epidermal Growth Factor Receptor Gene in Lung Cancer: Biological and Clinical Implications. Cancer Res 2004; 64: 8919-8923. [ Links ]
64. Kaneda H, Tamura K, Kurata T, Uejima H, Nakagawa K, Fukuoka M. Retrospective analysis of the predictive factors associated with the response and survival benefit of gefitinib in patients with advanced non-small-cell lung cancer. Lung Cancer 2004; 46:247-254. [ Links ]
65. Mitsudomi T, Kosaka T, Endoh H, et al. Mutations of the epidermal growth factor receptor gene predict prolonged survival after gefitinib treatment in patients with non-small-cell lung cancer with postoperative recurrence. J Clin Oncol 2005; 23(11):2513-20. [ Links ]
66. Ministry of Health, Labour and Welfare. Staff roster. January 10, 2005. (in Japanese). http://www.mhlw.go.jp/general/sosiki/kanbu/050110.html (accessed 2006 August 2) [ Links ]
67. Ministry of Health, Labour and Welfare. Minutes of Pharmaceutical Subcommittee II of the Pharmaceutical Affairs, Food, and Health Council. May 24, 2002. (in Japanese). http://www.mhlw.go.jp/shingi/2002/05/txt/s0524-2.txt (accessed 2006 August 2) [ Links ]
68. Fukushima M. Clinical trials in Japan. Nature Medicine 1995; 1 :12-13. [ Links ]
69. Fukushima M. The overdose of drugs in Japan. Nature 1989; 342: 850-851. [ Links ]
70. Ministry of Health, Labour and Welfare. Fukushima Masanori. Future directions for gefitinib use. March 7, 2005. (in Japanese). http://www.mhlw.go.jp/shingi/2005/03/dl/s0310-3i.pdf (accessed 2006 August 2) [ Links ]
71. Ministry of Health, Labour and Welfare. The states of the implement about approval conditions. January 20, 2005 (in Japanese). http://www.mhlw.go.jp/shingi/2005/01/dl/s0120-4c.pdf (accessed 2006 August 2) [ Links ]
72. Shepherd FA, Rodrigues Pereira J, Ciuleanu T, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123-32. [ Links ]
73. OSI Pharmaceuticals Inc. Label Tarceva® (erlotinib). October 18, 2005. http://www.fda.gov/medwatch/safety/2005/Nov_PI/Tarceva2_PI.pdf (accessed 2006 August 2) [ Links ]
74. Yamamoto N, Yamada Y, Shimoyama T, et al. A phase I study of erlotinib HCl in Japanese patients with various types of solid tumors. proc ASCO 2003; 22: 225. (abstract#903). [ Links ]
75. Tammaro KA, Baldwin PD, Lundberg AS. Interstitial lung disease following erlotinib (Tarceva) in a patient who previously tolerated gefitinib (Iressa). J Oncol Pharm Pract. 2005;11(3):127-30. [ Links ]

