










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
Print version ISSN 1137-6627
Anales Sis San Navarra vol.30 n.2 Pamplona May./Aug. 2007
Hipofunción glucocorticoide en la distrofia miotónica
Glucocorticoid hypofunction in myotonic dystrophy
L. Forga, E. Anda, F.J. Basterra, M.J. Goñi, F.J. Pineda
Servicio de Endocrinología. Hospital de Navarra. Pamplona
Dirección para correspondencia
RESUMEN
Introducción. La distrofia miotónica (DM1) es una enfermedad autonómica dominante cuyo defecto genético consiste en una expansión por repeticiones del triplete CTG en un gen que codifica una proteín-kinasa serina-treonina AMPc dependiente llamada DMPK. Se trata de una enfermedad multisistémica con conocida repercusión endocrinológica. En cuanto a la función suprarrenal, los resultados descritos han sido variables aunque últimamente se interpretan como indicadores de una hiperactividad del eje hipotálamo-hipófiso-adrenal.
Material y métodos. Se han estudiado 25 pacientes (13 hombres y 12 mujeres) afectos de DM1 a los que se ha analizado: cortisol y ACTH basales, test de estímulo con 0,25 mg de ACTH para cortisol y test de CRH para cortisol y ACTH. Asimismo se valoró el grado de expansión de CTG por Southern blot y PCR. Como grupo control para basales se estudiaron 25 individuos sanos equiparables por edad y sexo, a 11 de los cuales se realizó test de CRH.
Resultado. Se diagnosticó a un paciente de insuficiencia suprarrenal primaria no autoinmune. En el resto de casos no hubo diferencias entre la ACTH basal de pacientes y controles, y la respuesta de cortisol a ACTH fue normal. Los pacientes presentaron un nivel de cortisol basal más bajo (p<0,01) y también mostraron, tras estímulo con CRH, una menor respuesta de cortisol (p<0,05) con cifras medias de ACTH más elevadas.
Conclusiones. Nuestros datos difieren de las últimas publicaciones y apuntan a una hipofunción suprarrenal por falta de eficacia de la ACTH sobre su receptor o a nivel post-receptor. Sugerimos que la etiología puede estar relacionada con el defecto subyacente en el gen que codifica la DMPK.
Palabras clave. Distrofia miotónica. Test de CRH. Hipofunción glucocorticoide.
ABSTRACT
Introduction. Myotonic dystrophy (DM1) is an autosomal dominant disorder whose genetic defect consists of the amplification of an unstable CTG trinucleotide repeat in the 3 untranslated region of the dystrophia myotonica protein kinase gene (DMPK). This is a multi-systemic disease with a well-known endocrinological repercussion. With respect to the adrenal function variable results have been described, although lately they are interpreted as indicators of a hyperactivity of the hypothalamic-pituitary-adrenal (HPA) axis
Material and methods. Twenty-five patients (13 men and 12 women) with DM1 were recruited. They were analysed for: basal cortisol and ACTH, stimulus test with 0.25 mg of ACTH for cortisol and CRH test for cortisol and ACTH. Similarly, the degree of expansion of CTG was evaluated by Southern blot and PCR. Twenty-five healthy individuals, comparable by age and sex, were studied as a control group; the CRH test was carried out on 11 of them.
Result. One patient was diagnosed with primary non-autoimmune adrenal failure. In the rest of the cases there were no differences between the basal ACTH of patients and controls, and the cortisol response to ACTH was normal. The patients showed a lower level of basal cortisol (p<0.01) and also showed, following stimulation with CRH, a lower cortisol response (p<0.05) with higher average values of ACTH.
Conclusions. Our data differs from the latest publications and point to an adrenal hypofunction due to lack of efficacy of the ACTH on its receptor or at the post-receptor level. We suggest that the etiology might be related to the underlying defect in the gene that codifies DMPK.
Key words. Myotonic dystrophy. CRH test. Glucocorticoid hypofunction.
Introducción
La distrofia miotónica (designada DM1 por el Consorcio Internacional de Distrofia Miotónica) o enfermedad de Steinert es un alteración multisistémica de gran variabilidad clínica, heredada de forma autonómica dominante y con un índice de penetrancia muy elevado. Supone la forma heredada más frecuente de distrofia muscular entre adultos1. La causa genética reside en una expansión patológica del trinucleótido CTG situado en la región no transcrita 3 de un gen ubicado en el brazo largo del cromosoma 19 (19q13.3) que codifica para una proteín-kinasa (proteín kinasa de la distrofia miotónica: DMPK)2. El mecanismo por el cual la repetición expandida del DNA se traduce en sus manifestaciones clínicas y analíticas no se conoce3, aunque el número de repeticiones CTG se ha asociado con algunos datos clínicos y analíticos, especialmente disfunción cognitiva, hipogonadismo en el varón4 y respuesta incrementada de cortisol a ACTH tanto en hombres como en mujeres5.
Centrándonos en la función suprarrenal, se han descrito distintos hallazgos en los diferentes niveles del eje hipotálamo-hipófiso-adrenal (EHHA) que, en su mayoría apuntan a una hiperactividad. A. Pizzi y col fueron los primeros en hallar alteraciones en el ritmo circadiano de cortisol6. Sus resultados se ampliaron posteriormente al obtener niveles elevados de cortisol salivar3 y sérico y también de ACTH plasmática con ritmo circadiano aplanado7. En los estudios de estimulación con ACTH exógena, la respuesta de cortisol varía entre normalidad en todos los pacientes8, hiporespuesta en algún caso, sin alteración clínica6 e hiperrespuesta en presencia de largas expansiones de CTG5. No se han apreciado alteraciones en la respuesta del cortisol plasmático a la hipoglucemia insulínica8, a la metirapona9, a la CRH (estímulo directo10 o indirecto11) ni a la supresión con dexametasona3. Similar variabilidad ha ofrecido la respuesta de ACTH a las pruebas de estímulo, con resultados normales11 o aumentados a la hipoglucemia insulínica12, normales pero retrasados a la AVP exógena13 y elevados a la CRH ya fuera administrada directamente10 o estimulada indirectamente con naloxona11 o fenfluramina15.
Nuestro grupo publicó anteriormente el caso de un paciente afecto de síndrome pluriglandular no autoinmune (con anticuerpos negativos) que incluía insuficiencia suprarrenal primaria16. Con el presente trabajo hemos querido evaluar la función del EHHA en los pacientes con DM1 dados los discordantes hallazgos obtenidos en la literatura y teniendo como hipótesis que la alteración genética que subyace en la DM1 podría afectar a las hormonas que actúan por la vía de la proteína G (como la ACTH) y en la que diversas kinasas intervienen en el mecanismo de acción post-receptor, provocando hipofunción glandular.
Material y métodos
Sujetos estudiados. Se han estudiado 25 pacientes (13 hombres y 12 mujeres) diagnosticados de DM1 según los criterios de Jennekens y col17 (Tabla 1). La selección se realizó al azar, por orden consecutivo al que acudían a las consultas externas de especialidades de los hospitales: de Navarra, Virgen del Camino y Reina Sofía, y tras dar su consentimiento para la realización del estudio. La severidad de la alteración neuromuscular fue clasificada en tres grupos (de A a C, de menor a mayor gravedad) siguiendo los criterios de Novelli y col18. Como grupo control para las determinaciones basales se analizaron 25 voluntarios sanos, equiparados por edad y sexo. El test de estímulo con ACTH se valoró como normal o patológico según el criterio de normalidad universalmente aceptado: pico de cortisol 20 µg/dl19. El test de estimulación con CRH se realizó en 11 de los voluntarios sanos. Ninguno de los 50 individuos estudiados tomó medicación alguna durante al menos las 4 semanas previas a la realización de los análisis.

Protocolo del estudio. Los test de estimulación se practicaron de forma ambulatoria en días diferentes separados 72 horas. Los análisis se iniciaron a las 8 horas, tras un ayuno nocturno de 12 horas. El primer día se extrajo suero para determinar: cortisol y ACTH. A continuación se inyectaron 0,25 mg de Synacthen® diluido en 10 cc de suero fisiológico de forma ev lenta. Nuevas extracciones para cortisol a los 30 y 60 min. El segundo día de estudio se realizó el test de estímulo con CRH: determinación basal para cortisol y ACTH, inyección ev directa de 100 µg de CRH humana (Corticobiss®) y nuevas extracciones para cortisol y ACTH a los 15, 30, 60 y 90 min.
La muestra, una vez obtenida, se centrifugó y congeló de inmediato para evitar la degradación de ACTH
Los pacientes objeto de estudio no llevaban ningún tratamiento farmacológico capaz de alterar la función hipófiso-adrenal ni estaban afectos de enfermedad psiquiátrica alguna.
Determinaciones analíticas. El cortisol plasmático se midió por RIA (Sorin Biomedica, Vercelli, Italia). La sensibilidad del ensayo, con un IC de 95%, es de 10 ng/ml. Coeficiente de variación intraensayo 5%, Coeficiente de variación interensayo 4,9%.
La ACTH se midió por IRMA (Nichols Institute Diagnostics, San Juan Capistrano, USA). La sensibilidad del ensayo, con un IC de 95%, es de 1 pg/ml. Coeficiente de variación intraensayo 3%. Coeficiente de variación interensayo 6,8%.
Estudio genético. Se practicó en 16 pacientes que dieron su consentimiento. Se examinó el DNA mediante las técnicas de Southern blot y PCR (polymerase chain reaction). La técnica de Southern blot se utilizó para el análisis de expansiones largas y heterogéneas de CTG. El DNA fue digerido con las enzimas de restricción Eco RI o BgII, seguido de electroforesis en gel de agarosa al 0,8% y transferencia a membranas de nylon (Hybond N). El Southern Blot fue probado con el fragmento 1,8 kbp HindIII/BamHI de prueba pGB2.6 que detecta la expansión de las repeticiones. Posteriormente las muestras fueron lavadas en 0,5 mol/l de NaOH/1,5 mol/l NaCl, en 0,5 Tris (pH 7,0)/1,5 mol/l NaCl. Las amplificaciones de las repeticiones de CTG fueron medidas en el punto de mayor densidad.
La técnica de PCR fue usada para determinar el número de repeticiones de CTG en alelos normales y en amplificaciones de repeticiones pequeñas (<0,5 kb) en pacientes que no fueron resueltos por Southern blot. La PCR se llevó a cabo con los primers de oligonucleótidos 101 y 102 que flanquean las repeticiones de CTG. Los productos de PCR fueron resueltos por electroforesis en geles de azarosa Metaphor al 3,5%. Posteriormente el DNA fue transferido a membranas de nylon (Hybond N) y probado con un oligonucleótido (CTG)10 marcado con fósforo 32.
Para estudiar las posibles diferencias según el número de repeticiones, se ha utilizado la subdivisión entre <500 y 500 repeticiones del triplete en sangre periférica.
Datos y análisis estadísticos. Los resultados hormonales se expresan como la media ± la desviación estándar. Para el análisis de las diferencias entre controles y pacientes, y tras comprobar la ausencia de normalidad en las muestras, se ha empleado como prueba de comparación la U de Mann-Whitney. Para el análisis de las correlaciones entre las distintas variables, dado que no seguían una distribución normal, se usó el test de correlación de Spearman. En todos los casos se ha considerado significativo un valor de p<0,05. El paquete informático SPSS 13,0 ha sido utilizado para todos los análisis estadísticos.
Resultados
Los resultados obtenidos en los parámetros analizados se exponen en la Tabla 2. Al evaluar la función glucocorticoide basal, todos los pacientes resultaron clínica y analíticamente normofuncionantes excepto uno de ellos afecto de insuficiencia suprarrenal primaria no autoinmune que mostró atrofia de ambas suprarrenales en el TAC y requirió tratamiento hormonal sustitutivo. Los datos de ACTH basal no mostraron diferencias significativas entre pacientes y controles, a diferencia de los niveles de cortisol basal que fueron inferiores en los pacientes.

Salvo en el caso mencionado del paciente con insuficiencia suprarrenal, el resto mostraron respuesta normal de cortisol al estímulo con ACTH, siendo el pico medio de 30,47 ± 10,43 mg/dl, con un rango entre 12,80 y 53 mg/dl. La media de los puntos obtenidos durante esta prueba se representa en la figura 1.

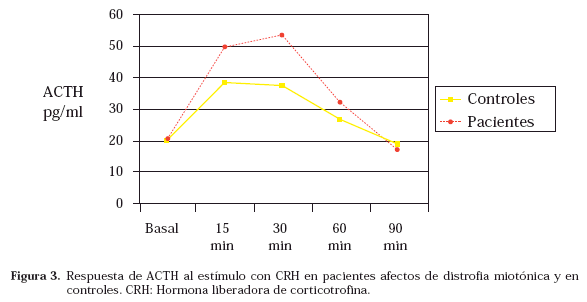
La respuesta al test de CRH mostró los datos de mayor interés. Para el análisis estadístico se excluyó al paciente afecto de insuficiencia suprarrenal primaria. En las figuras 2 y 3 se muestran respectivamente las medias de los valores de cortisol y ACTH en todos los puntos de la prueba. Los picos de respuesta de cortisol y ACTH a CRH se muestran en la tabla 2. No existió correlación entre la respuesta hormonal y la gravedad de la alteración muscular clínica ni con el número de repeticiones del triplete CTG en sangre periférica.

Discusión
Las últimas publicaciones sobre la función glucocorticoide en la DM1, apuntan a una hiperactividad del EHHA5,7. La causa radicaría en una mayor sensibilidad al estímulo por parte de la corteza adrenal, a un metabolismo anormal de los glucocorticoides (aumento de la actividad de la 11ßHSD1)5 o al efecto de la resistencia insulínica a través de sus mediadores20,21. Nuestros datos orientan en sentido contrario. Además de diagnosticar una insuficiencia suprarrenal primaria dentro de un síndrome pluriglandular no autoinmune, hemos observado un nivel de cortisol basal inferior y una menor respuesta de cortisol al estímulo con CRH acompañada de una respuesta mayor de ACTH, no significativa (posiblemente por la elevada desviación estándar, pero apreciable en la gráfica de respuesta) comparada con sujetos control. Este patrón sugiere una menor sensibilidad de la corteza suprarrenal al estímulo con ACTH que no se pone de manifiesto en respuesta al intenso estímulo agudo con 250 µg de ACTH ev, aunque sí se evidencia en condiciones basales y tras un estímulo más sutil como es la CRH. Jackson y col10 al practicar el test de estímulo con CRH ovina, obtuvieron también valores elevados de ACTH que no conllevaban aumento en la respuesta de cortisol y Grice y col11 hallaron idénticos resultados con la naloxona que actúa vía CRH. En nuestra opinión, y puesto que la CRH no actúa directamente sobre las suprarrenales, la menor respuesta de cortisol se debería a una menor eficacia de la ACTH tras interactuar con su receptor a nivel de corteza suprarrenal. Se trata de un receptor de superficie que presenta 7 dominios transmembrana y está ligado a la proteína Gs, activando a la adenilato ciclasa que da lugar al aumento del segundo mensajero cAMP, cuyo efector final es una proteín-kinasa A. El defecto subyacente en el gen que codifica la DMPK dependiente de AMPc podría ser la causa de la hiporespuesta adrenal ya que largas expansiones de CTG han sido encontradas en las suprarrenales22. Esta hipótesis ha sido también sugerida por otros autores23-25. Otra posible explicación sería una alteración del RNA del receptor de ACTH a nivel de corteza suprarrenal ya que parece existir una alteración general del RNA nuclear en la DM1. En la literatura médica figura otro caso de insuficiencia suprarrenal asociada a DM1, aunque en esta ocasión de origen autoinmune26. Dicha etiología no avala nuestra hipótesis aunque sí podría hacerlo la ya clásica publicación de Berthold y col27 en la que se demuestran diversos grados de atrofia suprarrenal en autopsias de pacientes afectos de DM1.
En conclusión, presentamos una serie de 25 pacientes afectos de DM1, a los que se ha practicado estudio de función suprarrenal glucocorticoide, habiéndose obtenido unos menores niveles de cortisol tanto basales como en respuesta a CRH a pesar de mediar cifras más elevadas de ACTH, lo que indicaría una menor eficacia a nivel de receptor o postreceptor. Estas diferencias halladas entre pacientes y controles, no necesariamente entrañan significado patológico.
Obviamente son necesarios más estudios para conocer cuál es la funcionalidad del EHHA en los pacientes afectos de DM1 y cuáles son los mecanismos implicados sobre todo dado que nuestros resultados difieren de lo últimamente publicado.
Bibliografía
1. Thornton C. The myotonic dystrophies. Semi Neurol1999; 19: 25-33. [ Links ]
2. Brook JD, McCurrah ME, Harley HG, Buckler AJ, Church D, Aburanti H et al. Molecular basis of myotonic dystrophy: expansion of a triplet (CTG) repeat at the 3 end of a transcript encoding a protein kinase family member. Cell 1992; 68: 799-808.
3. Johansson A, Henriksson A, Olofsson BO, Olsson T. Adrenal steroid dysregulation in dystrophia myotonica. J Intern Med 1999; 245: 345-351.
4. Mastrogiacomo I, Pagani E, Novelli G, Angelini C, Gennarelli M, Menegazzo E et al. Male hypogonadism in myotonic dystrophy is related to (CTG)n triplet mutation. J Endocrinol Invest 1994; 17: 381-383.
5. Johansson A, Andrew R, Forsberg H, Cederquist K, Walker BR, Olsson T. Glucocorticoid metabolism and adrenocortical reactivity to ACTH in myotonic dystrophy. J Clin Endocrinol Metab 2001; 86: 4276-4283.
6. Pizzi A, Fusi S, Forti G, Marconi G. Study of endocrine function in myotonic dystrophy. Ital J Neurol Sci 1985; 6: 457-467.
7. Johansson A, Carlström K, Ahrén B, Cederquist K, Krylborg E, Forsberg H et al. Abnormal cytokine and adrenocortical hormone regulation in myotonic dystrophy. J Clin Endocrinol Metab 2000; 85: 3169-3176.
8. Carter JN, Steinbeck KS. Reduced adrenal androgens in patients with myotonic dystrophy. J Clin Endocrinol Metab 1985; 60: 611-614.
9. Bernard-Weil E. Données neuro-endocriniennes nouvelles sur la maladie de Steinert particuliérment en ce qui concerne laxe hypothalamo-hypophyso-surrénalien et la régulation du métabolism phospho-calcique. Ann Endocrinol (Paris) 1972; 33: 251-266.
10. Jackson RV, Fleming GA, Sussman CR, Atkinson AB, Decherney GS, Debold CR et al. Increased proopiomelanocortin derived peptide release in myotonic dystrophy. Aus Paediatr J 1988; 24 (Suppl 1): 70-73.
11. Grice JE, Jackson RV, Hockings GI, Torpy DJ, Walters MM , Crosbie GV. Adrenocorticotropin hyperresponse to the corticotropin-releasing hormone-mediated stimulus of naloxone in patients with myotonic dystrophy. J Clin Endocrinol Metab 1995; 80: 179-184.
12. Mahler C, Parizel G. Hypothalamic pituitary function in myotonic dystrophy. J Neurol 1982; 226: 233-242.
13. Takase S, Okita N, Sakuma H, Mochizuki H, Ohara Y, Mizuno Y et al. Endocrinological abnormalities in myotonic dystrophy: consecutive studies of eight tolerance test in 26 patients. Tohoku J Exp Med 1987; 153: 355-374.
14. Grice JE, Jackson J, Hewett M, Penfold PJ, Jackson RV. Effect of exogenous arginine vasopressin on adrenocorticotropin and cortisol release in myotonic dystrophy patients: delayed responses of normal magnitude. J Neuroendocrinol 1991; 3: 65-68.
15. Grice JE, Jackson J, Penfold PJ, Jackson RV. Adrenocortocotropin hyperresponsiveness in myotonic dystrophy following oral fenfluramine administration. J Neuroendocrinol 1991; 3: 69-73.
16. Forga L, Rodríguez Erdozain RM, Menéndez EL, Anda E, Quesada Jiménez P. Insuficiencia suprarrenal primaria y atrofia pluriglandular en un paciente afecto de distrofia miotónica. Rev Neurol 1996; 24:91-93.
17. Jennekens FG, Kate LP, De Visser M, Wintzen AR. Diagnostic criteria for Duchenne and Becker muscular dystrophy and myotonic dystrophy. Neuromusc Dis 1991; 1: 389-391.
18. Novelli G, Gennarelli M, Menegazzo E, Mostacciuolo ML, Pizzuti A, Fattorini C et al. (CTG)n triplet mutation and phenotype manifestations in myotonic dystrophy patients. Biochem Med Metab Biol 1993; 50: 85-92.
19. May ME, Carey RM. Rapid adrenocorticotropic hormone test in practice. Retrospective review. Am J Med 1985; 79: 679-684.
20. Fernández-Real JM, Molina A, Broch M, Ricart W, Gutierrez C, Casamitjana R et al. Tumor necrosis factor system activity is associated with insulin resistance and dyslipidemia in myotonic dystrophy. Diabetes 1999; 48: 1108-1112.
21. Gómez JM, Molina A, Fernández-Castañer M, Casamitjana R, Martínez-Matos JA, Soler J. Insulin regulation of leptin synthesis and secretion in humans: the modelo of myotonic dystrophy. Clin Endocrinol (Oxf) 1999; 50: 569-575.
22. Kinoshita M, Takahashi R, Hasegawa T, Komori T, Nagasawa R, Hirose K et al. (CTG)n expansions in various tissues from a myotonic dystrophy patient. Muscle Nerve 1996; 19: 240-242.
23. Olsson T. Hyperkalaemia and selective hypoaldosteronism in myotonic dystrophy. Clin Endocrinol (Oxf) 2002; 56: 151-152.
24. Buyalos R, Jackson R, Grice GI, Hockings GI, Torpy DJ, Fox LM et al. Androgen response to hypothalamic-pituitary-adrenal stimulation with naloxone in women with myotonic dystrophy. J Clin Endocrinol Metab 1998; 83: 3219-3224.
25. Misra D, DeSilva S, Fellerman H, Dufour DR, Streeten DHP, Nylen ES. Hyperkalaemia and selective hypoaldosteronism in myotonic dystrophy. Clin Endocrinol (Oxf) 2002; 56: 271-275.
26. Pagliara S, Spagnuolo E, Ambrosone L, Barbato A, Tesauro P, Rambaldi M. Hyperthyroidism and Addisons disease in a patient with myotonic dystrophy. Arch Intern Med 1985; 145: 919-920.
27. Berthold H. Zur pathologischen anatomie der dystrophia myotonica (Curshmann-Steinert). Dtsch Z Nerv 1958; 178: 394.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Lluís Forga Llenas
Servicio de Endocrinología
Hospital de Navarra
C/Irunlarrea, 3
31007 Pamplona
Tfno. 848 42 20 38
Fax 848 42 20 38
E-mail: lforgall@cfnavarra.es
Aceptado para su publicación el 20 de noviembre de 2006.

