










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
Print version ISSN 1130-0108
Rev. esp. enferm. dig. vol.101 n.1 Madrid Jan. 2009
Oscillations in serum ferritin associated with antiviral therapy in chronic hepatitis C
Oscilaciones de la ferritina sérica asociadas al tratamiento antiviral en la hepatitis crónica por virus C
J. M. Ladero, G. López-Alonso, M. J. Devesa, F. Cuenca, L. Ortega1, M. Agreda, A. Suárez2, P. Ropero3 and M. Díaz-Rubio
Services of Gastroenterology (Liver Unit), 1Pathology, 2Clinical Microbiology, and 3Hematology. Hospital Clínico San Carlos. Universidad Complutense. Madrid, Spain
Supported in part with a grant by Schering-Plough.
ABSTRACT
Background: hyperferritinemia is often found in patients with chronic hepatitis C (CHC) and is predictive of poorer response to antiviral therapy.
Objective: to investigate changes in ferritinemia during and after antiviral therapy.
Patients and methods: serum ferritin levels were measured in 262 CHC patients (163 males, mean age 48.5 years ± 10.1) before and during antiviral therapy, and six months post-treatment in all 154 patients whit undetectable serum HCV-RNA after therapy completion.
Results: baseline serum ferritin was higher in patients with primary therapeutic failure than in those reaching sustained viral response (330 ± 291 ng/mL vs. 211 ± 192 ng/mL, p = 0.002). Serum ferritin transiently increased during therapy from baseline (257 ± 242 ng/mL vs. 875 ± 630 ng/mL, p < 0.001). Six months after finishing therapy, serum ferritin decreased under baseline values both in sustained responders (117 ± 102 ng/mL vs. 211± 192 ng/mL, p < 0.001) and, to a lesser extent, in relapsers (217 ± 174 ng/mL vs. 257 ± 221 ng/mL, p = 0.047).
Conclusions: baseline serum ferritin may predict response to antiviral treatment in chronic hepatitis C. Combined antiviral therapy induces a marked increase in serum ferritin that falls below baseline values after sustained viral response, suggesting that the cause of hyperferritinemia in many patients is HCV infection itself rather than iron overload.
Key words: Serum ferritin. Hepatitis C. Pegylated interferon. Ribavirin.
RESUMEN
Antecedentes: la hiperferritinemia es frecuente en los enfermos con hepatitis crónica C (HCC) y reduce las probabilidades de respuesta al tratamiento antiviral.
Objetivo: investigar las variaciones de la ferritina sérica durante y después del tratamiento y su relación con la respuesta al mismo.
Pacientes y métodos: la ferritina sérica se ha medido en 262 enfermos con HCC (163 hombres, edad media 48,5 años ± 10,1) antes y durante el tratamiento antiviral, y a los 6 meses de finalizado en los 154 enfermos con viremia indetectable al final del tratamiento.
Resultados: la ferritina sérica basal era más alta en enfermos con fracaso terapéutico primario que en los que consiguieron respuesta viral sostenida (RVS) (330 ± 291 ng/ml vs. 211 ± 192 ng/ml, p = 0,002). La ferritina sérica aumentó transitoriamente durante el tratamiento (257 ± 242 ng/ml vs. 875 ± 630 ng/ml, p < 0,001). La ferritina sérica descendió a valores inferiores a los basales seis meses después de finalizado el tratamiento en los pacientes con RVS (117 ± 102 ng/ml vs. 211± 192 ng/ml, p < 0,001) y, en menor grado, en los que sufrieron recidiva viral (217 ± 174 ng/ml vs. 257 ± 221 ng/m, p = 0,047).
Conclusiones: una ferritina sérica basal elevada se asocia con mayor riesgo de fracaso terapéutico en la HCC. El tratamiento antiviral induce un marcado incremento de la ferritina sérica que vuelve a valores por debajo de los basales en los enfermos que obtienen R vs. Esto sugiere que la causa de hiperferritinemia en la mayoría de los enfermos es la propia infección por VHC y no la sobrecarga de hierro.
Palabras clave: Ferritina sérica. Hepatitis C. Interferón pegilado. Ribavirina.
Introduction
Liver iron overload is considered a surrogate marker of liver fibrosis (1,2), and is associated with impaired response to interferon-based therapy (3,4). However, the frequency and severity of iron overload in chronic hepatitis C, which oscillates between broad limits, from 7 to 32% (2,5-8), remains controversial. Iron accumulation in the liver may aggravate tissue damage by generating free radicals, releasing proinflammatory and profibrogenic cytokines, and interfering with the immune system (9,10).
High serum ferritin is a common finding in many acquired liver diseases, particularly in chronic hepatitis C (CHC), alcoholic liver disease, and non-alcoholic fatty liver disease (11,12). Ferritin is the iron-storage protein in tissues, and its serum levels correlate with total iron content; thus, it may be speculated that high serum ferritin levels may be a marker of iron overload. This is usually true in the absence of chronic inflammation, with very rare exceptions (13), but ferritin synthesis and release are increased in states of chronic immune stimulation (9,14), and the actual meaning of hyperferritinemia in CHC has not been fully elucidated. If it were a consequence of hepatic iron overload, a positive correlation between both parameters would be expected. However, this is only true when CHC coincides with a genetic cause of iron overload, as it occurs in carriers of HFE gene mutations (mainly the C282Y mutation) or other genes related to iron homeostasis (6,8,15). Contrarily, if elevated serum ferritin in CHC were a surrogate marker of a chronic inflammatory state, we should expect that serum ferritin levels would return to normal values after successful antiviral therapy.
The aim of this work was to study the variations of serum ferritin concentrations during and after antiviral therapy in patients with chronic hepatitis C virus (HCV) infection, and to investigate whether a potential association with therapeutic response exists.
Patients and methods
Since January 2004, serum ferritin concentration is systematically determined in all CHC patients seen at our Unit before initiating treatment with pegylated interferon (α2a or α2b) and ribavirin, and at regular intervals during therapy. Serum ferritin is also determined in patients who reach a final virological response (defined as blood HCV-RNA undetectable at the end of therapy) 24 weeks after finishing treatment. We reviewed the clinical records of these patients with the aim of studying variations in ferritinemia during and after therapy. Patients were classified in four groups after excluding those who dropped out for voluntary discontinuation -- group 1: sustained viral response (SVR); group 2: primary failure of therapy; group 3: early discontinuation due to severe intolerance; and group 4: relapse after transient viral response. Other exclusion criteria were chronic hemolysis, previous therapy with iron, coinfection with HIV, and active infection with HBV. HFE genotype was not systematically determined in patients with elevated serum ferritin, but only in some cases according to the clinical criteria of the attending physician. All patients were submitted to the current standard of care on an outpatient basis. The study protocol was approved by the Local Ethics Committee and conforms to the ethical guidelines of the Declaration of Helsinki.
The diagnosis of chronic hepatitis C was based on the results of clinical evaluation and a combination of biochemical and viral tests. All patients were positive for anti-HCV antibody and for HCV RNA at the time when antiviral therapy was started. A quantitative analysis of HCV-RNA was performed with the Cobas Amplicor HCV Monitor version 2.0 (Roche Molecular Diagnostic). The lower limit of detection was 600 IU/mL, and the upper level was 8.5 x 105 IU/mL. After July 2005, viral RNA was extracted automatically using Cobas AmpliPrep, but viral load was detected using Real-Time PCR Cobas TaqMan (Roche Diagnostics), that has a dynamic range between 10 IU/mL and 2 x 108 IU/mL.
HCV genotypes were determined using a reverse hybridization assay (INNO-LiPA. Innogenetics), based on the variations found in the 5' untranslated region of various hepatitis C virus sequences, after amplification by a reverse transcription polymerase chain reaction (RT-PCR). Active hepatitis B virus (HBV) infection and HIV infection were excluded by standard serological analysis. Active infection with HBV and HIV were excluded using standard serological methods. Hematological and biochemical parameters were determined with standard methods in the clinical laboratory.
The statistical analysis was performed using Student's t-test or the Mann-Whitney U-test, as indicated, to compare continuous variables, odds ratios with 95% confidence intervals for categorical variables, and Spearman's rank test to estimate correlations between variables. Values are expressed as mean ± standard deviation (SD) or as percentages. All tests were two-sided. Calculations were made using the SPSS statistical software package for Windows (version 15.0; SPSS) and Epi-Info 2002 (Centers for Disease Control and Prevention). The null hypothesis was rejected when p < 0.05.
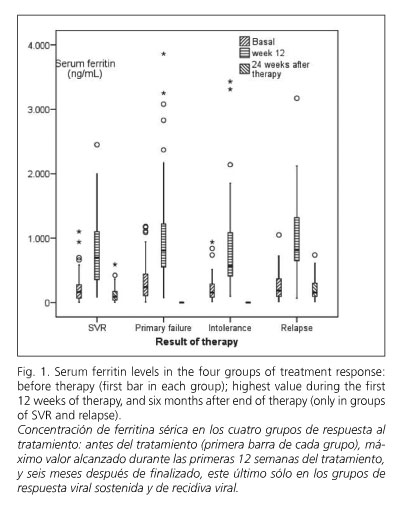
Results
Two hundred and sixty-two patients (163 male; mean age 48.5 years ± 10.1) fulfilling the inclusion criteria were considered for the analysis. One hundred and eighty-three were infected with HCV genotype 1 (143 genotype 1b), 55 with genotype 3, and the remaining with genotypes 2, 4 and 5. Pre-treatment liver biopsies were available for 136 patients, and were evaluated according to Knodell et al. (16) by the same pathologist. Perls iron specific staining was systematically performed. Table I reflects the biochemical, virological and histological data of the whole series and of the four response groups.
Baseline serum iron concentrations over 150 μγ/ml were detected in 27.8% of patients. Baseline ferritinemia was elevated in 25.2% of male patients (> 350 ng/ml) and in 19.2% of female patients (> 250 ng/ml). Table II shows the mean values of serum ferritin before and during therapy in the whole group and in the subgroups according to response to therapy, as well as those obtained 24 weeks after therapy completion in groups 1 (SVR) and 4 (relapse). Baseline serum ferritin was significantly higher in all 81 patients (group 2) with primary therapeutic failure than in the 121 patients (group 1) who reached SVR (330 ± 291vs. 211 ± 192, p = 0.002). No significant differences in baseline serum ferritin were observed between groups 1 (SVR), 3 (intolerance) and 4 (relapse), nor in baseline serum iron concentrations between response groups.
Baseline serum ferritin correlated at a significant level (p ≤ 0.001) with the following baseline values: body weight, hemoglobin, total serum bilirubin, serum ALT, serum AST, and serum iron.
Mean serum ferritin increased during the first 12 weeks of therapy from 257 ± 242 ng/ml to 875 ± 630 ng/dl (p < 0.001), to diminish slowly during the remaining part of therapy (613 ± 418 ng/dl at week 24 in the 179 patients with available values). Serum ferritin increased during therapy in 259 of the 262 patients included in the study. The median peak ferritin level reached during therapy was lower in the SVR group than in the other three groups, a difference that reaches statistical significance when comparing SVR and primary failure groups (p = 0.018).
The highest serum ferritin level reached between weeks 2 and 12 of therapy correlated at a significant level (p ≤ 0.001) with the following parameters: baseline body weight; baseline serum iron; platelet count and total serum bilirubin at week 2, and serum ALT, serum AST and serum GGT at weeks 2, 6 and 12. No significant relation exists between the highest serum ferritin value reached during therapy and hemoglobin values at weeks 2, 6 and 12 of therapy, nor with the baseline viral load considered as a dichotomous (low vs. high) variable (low ≤ 400.000 IU) (17).
As it is shown in table II, serum ferritin value 24 weeks after therapy completion was significantly lower than baseline figures in the 121 patients who obtained sustained viral response. This parameter actually decreased in 88.4% of patients included in this group. This difference is much smaller in the 33 patients who experienced virological relapse, in whom serum ferritin decreased only in 66.7%. This difference between both response groups correlates well with the evolution of serum ALT, which fell from 110 ± 88 U/L to 20 ± 9 U/L in sustained responders (p < 0.001) with only two exceptions, and from 89 ± 68 U/L to 57 ± 40 U/L in relapsers (p = 0.033), with an actual decrease in 75.8% of patients.
When the same analysis was applied separately to the 183 genotype 1 patients, the results were very close to those found in the whole group, although the drop in serum ferritin 24 weeks after end of therapy in relapsers did not reach statistical significance (data not shown).
In the 136 patients with available liver biopsy there is a non-significant although progressive increase in baseline serum ferritin according to fibrosis stage in the Knodell score: stage 0 (20 patients) = 104 ± 159 ng/mL; stage 1 (59 patients) = 212 ± 158 ng/mL; stage 3 (44 patients) = 260 ± 274 ng/mL; stage 4 (13 patients) = 408 ± 396 ng/mL (p stage 0 vs. stage 4 = 0.197).
Stainable iron was detected in the liver biopsy of 8 patients (5.90% of cases with liver biopsy). Baseline serum ferritin was higher in these patients than in the remaining 128 with liver biopsy (481 ± 450 ng/dl vs. 231 ± 210 ng/ml, p = 0.083). No significant differences were observed for baseline serum iron values, nor for the maximal ferritin value during the first 12 weeks of therapy.
HFE genotype is available in 37 patients in whom the attending physician considered that serum iron, ferritin and/or transferrin saturation index justified this study. There were 6 single heterozygotes for the C282Y mutation, 12 single heterozygotes for the H63D mutation, 1 composed C282/H63D heterozygote, 1 H63D homozygote and 17 wild type genotypes. No differences in baseline serum iron, baseline ferritinemia and in highest serum ferritin value reached during the first 12 weeks of therapy were observed between carriers and non-carriers of HFE mutations. A liver biopsy was available in 24 of these patients, with stainable iron in 5, three of them being carriers of one HFE mutation. Fifteen of these 37 patients obtained SVR (three C282Y carriers, five H63D carriers and seven patients without HFE gene mutations).
Discussion
In this study we observed an impressive and nearly universal elevation of serum ferritin during combined antiviral therapy for CHC. The maximal values reached are slightly higher in the groups of primary failure, relapse, and intolerance than in the SVR group, but the increases are proportional to baseline values and the slopes are no different. The mechanism of hyperferritinemia is probably related to the immunostimulant and immunoregulatory effects of IFN-α2 through its attachment to cell-surface receptors and activation of multiple interferon-stimulated genes (18,19), and to the induction of a Th-1 response that increases the synthesis and liberation of proinflammatory cytokines (i.e. TNF-α, IL-1 and IL-6) (20), which are known up-regulators of the synthesis of ferritin (21). Stam et al. (22) have shown a significant increase in serum ferritin in a group of patients treated with high doses of interferon α after the surgical excision of a melanoma. Recently, Ferrara et al. (23) reported, in abstract form, a study with results quite similar to ours with regard to a marked rise of serum ferritin during therapy, although they do not provide any data on its subsequent evolution.
Although serum ferritin is a well-known acute phase reactant, other possible causes for hyperferritinemia during antiviral therapy should be discussed. Ribavirin induces hemolysis, and the increase in serum ferritin may be due to accelerated iron turnover. On the other side, serum bilirubin may increase because of heme catabolism. In this study, baseline values for hemoglobin and total bilirubin correlated significantly with baseline serum ferritin, and this correlation persisted with serum bilirubin at week two of therapy, but not thereafter, and in no moment was that detected with hemoglobin. If hemolysis had been causally related to increased serum ferritin, narrowed, more significant correlations with hemoglobin and bilirubinemia should be expected during therapy versus baseline.
Our data on HFE gene mutations are very limited and biased through patients with higher baseline serum iron and ferritin values, but they do not confirm previous data on a possible association between carrier status for HFE gene mutations and an increased chance of obtaining SVR (24,25), a question not confirmed in a recent review (26).
Our results are in accordance with previous findings (23,24,27) indicating that high baseline serum ferritin levels are predictors of poor response to combined antiviral therapy for CHC. This suggests the convenience of analyzing whether ferritinemia may improve the multivariate predictive models aimed at establishing the individual chance of reaching therapeutic success in patients with CHC. Such analysis was not an aim of the present study, and should be based on a sample of greater size. Table I confirms that many of the reported factors predicting a primary therapeutic failure (older age, viral genotype 1, lower platelet count, high viral load, higher AST and GGT levels, and lower cholesterol, and more advanced fibrosis in liver biopsy) are more prevalent in the group of primary failure than in the group with SVR.
From a practical point of view, we think that the most interesting finding of this study is that serum ferritin falls not only to normal values, but as low as half its baseline figures at 24 weeks after therapy completion in patients who obtained SVR. Meanwhile, patients with virological relapse showed a much more modest decrease in serum ferritin. This observation leads to think that increased levels of serum ferritin in non-treated CHC are most often a consequence of actions by HCV itself or of chronic inflammation, which stimulates immune response. This conclusion is supported by our observation that serum ferritin levels parallel serum ALT, which is considered a biochemical marker of necroinflammation in the liver. Iron overload should be a secondary event limited to patients with associated specific conditions (i.e., carriers of HFE gene mutations or heavy drinkers).
Acknowledgements
The authors thank Dr. Julio Mayol for his critical revision of the manuscript.
References
1. Metwally MA, Zein CO, Zein NN. Clinical significance of hepatic iron deposition and serum iron values in patients with chronic hepatitis C infection. Am J Gastroenterol 2004; 99: 286-91. [ Links ]
2. Guyader D, Thirouard AS, Erdtmann L, Rakba N, Jacquelinet S, Danielou H, et al. Liver iron is a surrogate marker of severe fibrosis in chronic hepatitis C. J Hepatol 2007; 46: 587-95. [ Links ]
3. Shedlofsky SI. Role of iron in the natural history and clinical course of hepatitis C disease. Hepatogastroenterology 1998; 45: 263-9. [ Links ]
4. Eisenbach C, Gehrke SG, Stremmel W. Iron, the HFE gene, and hepatitis C. Clin Liver Dis 2004; 8: 775-85. [ Links ]
5. Distante S, Bjoro K, Hellum KB, Myrvang B, Berg JP, Skaug K, et al. Raised serum ferritin predicts non-response to interferon and ribavirin treatment in patients with chronic hepatitis C infection. Liver 2002; 22: 269-75. [ Links ]
6. Ladero JM, Ropero P, Ortega L, Taxonera C, González FA, López-Alonso G et al. HFE gene mutations, hepatic iron content, and histological severity in hepatitis C virus-induced chronic hepatitis. Rev Esp Enferm Dig 2003; 95: 833-6. [ Links ]
7. Silva IS, Perez RM, Oliveira PV, Cantagalo MI, Dantas E, Sisti C, et al. Iron overload in patients with chronic hepatitis C virus infection: clinical and histological study. J Gastroenterol Hepatol 2005; 20: 243-8. [ Links ]
8. Valenti L, Pulixi EA, Arosio P, Cremonesi L, Biasiotto G, Dongiovanni P, et al. Relative contribution of iron genes, dysmetabolism and hepatitis C virus (HCV) in the pathogenesis of altered iron regulation in HCV virus hepatitis. Hematologica 2007; 92: 1037-42. [ Links ]
9. García-Buey ML, González F. Iron overload and chronic hepatitis C: the role of HFE gene mutations. Rev Esp Enferm Dig 2003; 95: 824-8. [ Links ]
10. Weiss G. Modification of iron regulation by inflammatory response. Best Pract Res Clin Haematol 2005; 18: 183-201. [ Links ]
11. Di Bisceglie AM, Axiotis CA, Hoofnagle JH, Bacon BR. Measurements of iron status in patients with chronic hepatitis. Gastroenterology 1992; 102: 2108-13. [ Links ]
12. Shan Y, Lambrecht RW, Bomkovsky HL. Association of hepatitis C virus infection with serum iron status: Analysis of data from the third National Health and Nutrition Examination Survey. Clin Infect Dis 2005; 40: 834-41. [ Links ]
13. Cazzola M. Role of ferritin and ferroportin genes in unexplained hyperferritinemia. Best Pract Res Clin Haematol 2005; 18: 251-63. [ Links ]
14. Janka GE. Hemophagocytic syndrome. Blood Rev 2007; 21: 245-53. [ Links ]
15. Erhardt A, Maschner-Olberg A, Mellenthin C, Kappert G, Adams O, Donner A, et al. HFE mutations and chronic hepatitis C: H63D and C282Y heterozygosity are independent risk factors for liver fibrosis and cirrhosis. J Hepatol 2003; 38: 335-2. [ Links ]
16. Knodell RG, Ishak KG, Black WC, Chen TS, Craig R, Kaplowitz N, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology 1981; 1: 431-5. [ Links ]
17. Zeuzem S, Fried MW, Reddy KR, Marcellin P, Diago M, Craxi A, et al. Improving the clinical relevance of pre-treatment viral load as a predictor of sustained virological response (SVR) in patients infected with hepatitis C genotype 1 treated with peginterferon alfa-2a (40 kDa) (PEGASYS) plus ribavirin (COPEGUS). Hepatology 2006; 44: 267A. [ Links ]
18. Hoofnagle JH, Seeff LB. Peginterferon and ribavirin for chronic hepatitis C. N Engl J Med 2006; 355: 2444-51. [ Links ]
19. Chevaliez S, Pawlotsky J-M. Interferon-based therapy of hepatitis C. Adv Drug Deliv Rev 2007; 59: 1222-41. [ Links ]
20. Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood 2002; 99: 3505-16. [ Links ]
21. Tilg H, Kaser A. Type I interferons and their therapeutic role in Th2-regulated inflammatory disorders. Expert Opin Biol Ther 2004; 4: 469-81. [ Links ]
22. Stam TC, Swaak AJ, Kruit WH, Eggermont AM. Regulation of ferritin: a specific role for interferon-alpha (IFN-alpha)? The acute phase response in patients treated with IFN-alpha-2b. Eur J Clin Invest 2002; 32(Supl. 1): 79-83. [ Links ]
23. Ferrara F, Guido M, Ventura P, Geetti A, Abbati G, Corradini E, et al. Antiviral treatment profoundly affects iron status in HCV patients: implications for management and treatment outcome. J Hepatol 2006; 46(Supl. 1): S222. [ Links ]
24. Lebray P, Zylberberg H, Hue S, Poulet B, Carnot F, Martin S, et al. Influence of HFE gene polymorphisms on the progression and treatment of chronic hepatitis C. J Viral Hepat 2004; 11: 175-82. [ Links ]
25. Bonkovsky HL, Naishadham D, Lambrecht RW, Chung RT, Hoefs JC, Nash SR, et al. Roles of iron and HFE mutations on severity and response to therapy during retreatment of advanced chronic hepatitis C. Gastroenterology 2006; 131: 1440-51. [ Links ]
26. Gattoni A, Parlato A, Bangieri B, Bresciani M, Derna R, Baldassare R. Role of hemochromatosis genes in chronic hepatitis C. Clin Ter 2006; 157: 61-8. [ Links ]
27. Hofer H, Österreicher C, Jessner W, Penz M, Steindl-Munda P, Wrba F, et al. Hepatic iron concentration does not predict response to standard and pegylated-IFN/ribavirin therapy in patients with chronic hepatitis C. J Hepatol 2004; 40: 1018-22. [ Links ]
![]() Correspondence:
Correspondence:
José M. Ladero.
Servicio de Gastroenterología (Unidad de Hepatología).
Hospital Clínico San Carlos.
C/ Doctor Martín Lagos, s/n.
28040 Madrid, Spain.
e-mail: jladero.hcsc@salud.madrid.org
Received: 17-10-08.
Accepted: 09-12-08.

