










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkFarmacia Hospitalaria
versión On-line ISSN 2171-8695versión impresa ISSN 1130-6343
Farm Hosp. vol.44 no.3 Toledo may./jun. 2020 Epub 19-Oct-2020
https://dx.doi.org/10.7399/fh.11280
Revisión
Biosimilares de anticuerpos monoclonales en enfermedades inflamatorias y cáncer: situación actual, retos y oportunidades
1Servicio de Farmacia, Hospital Universitario Virgen Macarena, Sevilla. España.
2Servicio de Farmacia, Hospital Clínico Universitario San Carlos, Madrid. España.
3Amgen S.A., Barcelona, España.
El proceso de aprobación de los biosimilares de anticuerpos monoclonales en la Unión Europea está dirigido a descartar la presencia de diferencias significativas con el biológico original en los atributos de calidad, eficacia, inmunogenicidad y seguridad. Proporciona además la justificación para extrapolar la evidencia obtenida con un biosimilar en al menos una indicación al resto de indicaciones aprobadas para su biológico original, simplificando el programa de desarrollo de los biosimilares. Los biosimilares de anticuerpos monoclonales disponibles en la Unión Europea para el tratamiento de enfermedades inflamatorias y del cáncer han cumplido todos los requerimientos establecidos para la aprobación, y en muchos casos disponen de evidencia adicional. Además, los datos de uso en la vida real están confirmando la seguridad y eficacia de estos fármacos en las distintas patologías en las que se están utilizando. En España, varias sociedades médicas avalan el proceso regulatorio de los biosimilares y reconocen su papel en la eficiencia del sistema sanitario. No obstante, todavía existen algunas barreras que limitan su uso. La aplicación de diferentes medidas a nivel de paciente, prescriptor, institucional y nacional podría aumentar la penetración de los biosimilares, liberando recursos que podrían invertirse en otras terapias y, potencialmente, favorecer la innovación.
PALABRAS CLAVE: Biosimilares; Extrapolación; Intercambiabilidad; Eficiencia; Nocebo; Enfermedades inflamatorias; Cáncer; España
Introducción
Un biosimilar es un fármaco que contiene una versión del principio activo de un fármaco biológico original (también llamado “producto de referencia")1. El proceso de producción de los fármacos biológicos es complejo y sujeto a múltiples variables, ya que implica a organismos vivos y con frecuencia tecnología de ADN recombinante. Como resultado, los propios biológicos originales presentan variabilidad entre diferentes lotes de fabricación, e incluso dentro de un mismo lote, que se debe mantener dentro de unos márgenes aceptables para evitar un impacto en los resultados clínicos. Las compañías farmacéuticas involucradas en el desarrollo de biosimilares, además, no tienen acceso a las especificaciones de fabricación de los biológicos originales (por ser información confidencial). Por tanto, estas compañías deben diseñar sus propios procedimientos, y mejorarlos hasta que los atributos críticos de sus biosimilares (aquellos que pueden afectar a la farmacocinética (FC), eficacia y seguridad) están dentro de un rango acceptable2. En la práctica, esto significa que, aunque un biosimilar nunca puede ser una copia exacta del biológico original, debe ser altamente similar en cuanto a los atributos críticos. Una vez se ha conseguido esto, el biosimilar debe pasar por un proceso de aprobación específico para confirmar la ausencia de diferencias clínicamente significativas respecto al biológico original.
El presente trabajo tiene por objetivo describir la situación actual de los biosimilares de anticuerpos monoclonales (AcMs) en la Unión Europea (UE) y en España. En primer lugar, revisamos el proceso regulatorio de los fármacos biosimilares en la UE, poniendo especial énfasis en las particularidades de los AcMs debido a su complejidad. Este proceso proporciona la justificación para la extrapolación y el switch, dos de los aspectos más controvertidos de los biosimilares, que se discuten en mayor profundidad. Seguidamente, describimos los biosimilares de AcMs actualmente disponibles en la UE en dos áreas terapéuticas en las que se usan ampliamente (enfermedades inflamatorias y oncología). Analizamos brevemente el diseño de los ensayos pivotales que han llevado a su aprobación, los aspectos que apoyan la extrapolación, y la evidencia disponible sobre otros puntos relevantes (datos a largo plazo, switch). Finalmente, nos centramos en la situación actual y las perspectivas de futuro de los biosimilares en España, extensivas a los biosimilares de AcMs, y exponemos las medidas que podrían favorecer el uso de estos fármacos, contribuyendo a la eficiencia del sistema sanitario.
Regulación de los biosimilares en la Unión Europea
Los fármacos biológicos (incluyendo los AcMs) deben pasar por un proceso de aprobación centralizado antes de poder ser comercializados en la UE. El proceso específico para los biosimilares se instauró en 2004, y su objetivo es confirmar la ausencia de diferencias clínicamente significativas respecto al biológico original. Esto se consigue mediante estudios comparativos, que se realizan de forma escalonada, de manera que los resultados obtenidos en cada fase determinan los estudios requeridos en la siguiente3. La Figura 1 muestra el peso relativo de cada tipo de evidencia en el proceso de aprobación de los fármacos biológicos y sus biosimilares.
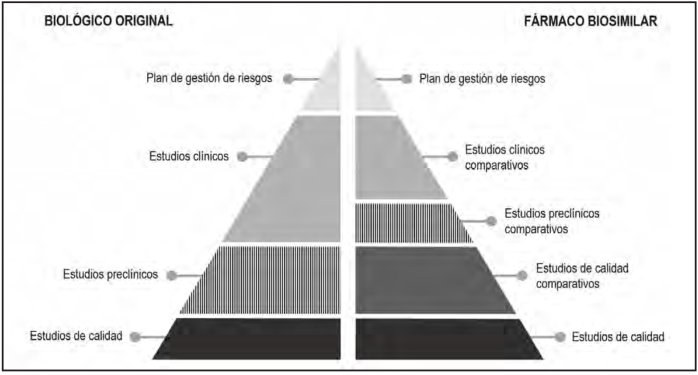
Figura 1. Requerimientos para la aprobación: diferencias entre los fármacos biológicos originales y biosimilares.
El primer paso consiste en estudios de calidad, ya que se consideran mucho más sensibles que los ensayos clínicos para detectar diferencias menores que pueden tener un impacto en la seguridad, eficacia e inmunogenicidad. Los estudios de calidad pueden implicar de 20 a 40 pruebas analíticas4, que comparan las estructuras primarias, modificaciones postraduccionales, variantes, estructuras de orden superior y actividad biológica del biosimilar y el biológico original. En el caso de los AcMs, que tienen múltiples dominios funcionales, caracterizar la actividad biológica implica no solo determinar el mecanismo de acción, sino también la función de los fragmentos de unión al antígeno (Fab) y cristalizable (Fc) por separado y en conjunto. Finalmente, se analizan las impurezas, la formulación, la potencia y la estabilidad2.
El siguiente paso son los estudios preclínicos que, en el caso de los AcMs, siempre deben incluir estudios in vitro de farmacodinámica (FD). En ellos se compara la unión de las regiones Fab y Fc de ambos productos (biosimilar y original) a sus moléculas diana, y las funciones mediadas por esta unión. Si los estudios de calidad han mostrado diferencias relevantes respecto al biológico original, si el biológico original media efectos que no pueden ser completamente elucidados mediante estudios in vitro (lo que ocurre con varios AcMs), o si persiste cualquier otro tipo de incertidumbre, se requieren estudios in vivo (FC, FD y/o seguridad) antes de proceder a los estudios clínicos. Los estudios in vivo se deben realizar en una especie (generalmente primates, por la especificidad de los AcMs) y/o modelo (por ejemplo, ratones transgénicos, modelos de xenoinjerto) relevante; si no se dispone de ellos, el desarrollador del biosimilar puede proceder directamente a la fase clínica, siempre que se apliquen medidas para mitigar los potenciales riesgos5.
Dado que el biológico original ya ha demostrado eficacia, seguridad y un perfil beneficio/riesgo positivo, el objetivo principal de la fase clínica del biosimilar es únicamente demostrar comparabilidad con el biológico original. El tipo de estudios a realizar dependerá de la complejidad de la molécula. En general, para todos los biosimilares se requieren estudios de FC/FD e inmunogenicidad3. Los estudios de FD únicamente se deben realizar si existe un marcador subrogado de eficacia válido, lo cual no siempre es el caso para los AcMs. Por otro lado, los datos clínicos de FC son especialmente relevantes. Si el biosimilar demuestra tener una exposición similar a la del biológico original, junto con datos analíticos y funcionales favorables, el desarrollador puede realizar directamente estudios clínicos de fase III a la misma dosis aprobada para el AcM original, sin tener que realizar antes estudios de fase II2.
Para algunos biosimilares, si existen marcadores clínicos válidos de FD, no es necesario realizar estudios clínicos más allá de la fase I. Los AcMs, no obstante, son moléculas especialmente complejas. Por tanto, por robusta que sea la evidencia recogida en los estudios de FC/FD, se impone realizar como mínimo un ensayo de fase III de equivalencia en eficacia, que además evalúe la seguridad3. En estos estudios de fase III, las poblaciones y las variables escogidas deben ser lo suficientemente sensibles para detectar diferencias entre el biológico original y el biosimilar, en caso de que existan. Estas variables no siempre coinciden con las utilizadas habitualmente en la indicación/área terapéutica escogida. Por ejemplo, en oncología, la tasa de respuesta se considera una variable adecuada para evaluar la equivalencia entre los biosimilares y los biológicos originales, si es suficientemente sensible a la acción de los fármacos y no está influenciada por factores externos5. El posicionamiento de la Sociedad Española de Oncología Médica (SEOM) sobre anticuerpos biosimilares se muestra de acuerdo con esto; pero también indica que, en los estudios con biosimilares, sería deseable incluir variables tradicionales de eficacia como la supervivencia libre de progresión o la supervivencia global6.
El tema de la inmunogenicidad merece una atención especial. Todos los productos biológicos tienen una capacidad intrínseca para desencadenar reacciones inmunes no deseadas, y los AcMs no son una excepción. Dado que no son tratamientos sustitutivos, los AcMs no suelen desencadenar la producción de anticuerpos neutralizantes contra moléculas endógenas (como puede ser el caso de las eritropoyetinas recombinantes)7. Aun así, una respuesta inmune al AcM puede reducir o eliminar la respuesta clínica, o desencadenar reacciones adversas graves8. Por tanto, para ser aprobados en la UE, los AcMs biosimilares deben demostrar que no presentan diferencias significativas de inmunogenicidad respecto a sus biológicos originales.
El primer paso para determinar la inmunogenicidad de un biosimilar es caracterizar mediante técnicas analíticas los factores relacionados con el fármaco que están implicados en el desarrollo de anticuerpos antifármaco (secuencia de aminoácidos, glicosilación, formulación e impurezas) y compararlos con los del biológico original3,8,9. No obstante, el desarrollo de anticuerpos también depende de factores relacionados con el paciente (por ejemplo, edad, situación del sistema inmune), con la enfermedad (como comorbilidades, tratamientos concomitantes) y con el tratamiento en estudio (por ejemplo, exposición)1,3. Además, en el caso de los AcMs es particularmente difícil predecir las potenciales reacciones inmunológicas en humanos partiendo únicamente de las diferencias a nivel de producto. Los ensayos con animales tampoco son especialmente sensibles a este respecto. Por tanto, la aprobación de un AcM biosimilar siempre requerirá de datos clínicos de inmunogenicidad, que se pueden obtener durante los estudios de FC o de eficacia/seguridad, o de forma aislada. A la hora de escoger la población, se ha de tener en cuenta que los pacientes sanos (que generalmente participan en estudios de FC) pueden ser la población más sensible para detectar diferencias en la inmunogenicidad, por presentar una respuesta inmune más fuerte y rápida1,3.
Una vez se han obtenido los datos descritos anteriormente, el ultimo paso para optar a la aprobación del biosimilar es el mismo que para los biológicos originales: presentar un plan de gestión de riesgos3. Este incluye un plan de farmacovigilancia y medidas de minimización de riesgos, y se basa en la experiencia ganada con el biológico original3. Como parte del plan de gestión de riesgos, durante los cinco primeros años tras la aprobación, la ficha técnica y el prospecto deben incluir un triángulo negro invertido, acompañado de un texto que invita a los profesionales sanitarios y pacientes a notificar las reacciones adversas que puedan ocurrir, para indicar que el medicamento está sujeto a un seguimiento especialmente intensivo. El triángulo negro se aplica a todos los fármacos biológicos (no únicamente a los biosimilares) que han sido aprobados tras demostrar un perfil riesgo/ beneficio favorable. El objetivo del seguimiento es recopilar información que no se ha podido obtener durante el desarrollo (por ejemplo, efectos a largo plazo) y asegurar que el perfil de seguridad sigue siendo favorable10. La legislación europea -y por extensión, la española- obliga a incluir el nombre comercial (unívoco respecto al nombre del principio activo) y el número de lote en las notificaciones de reacciones adversas que impliquen biosimilares, para facilitar la trazabilidad11,12. No obstante, la introducción de esta legislación no ha aumentado la inclusión de nombres comerciales en las notificaciones de seguridad relacionadas con fármacos biológicos, y la inclusión de los números de lote continúa siendo muy baja (5-21%)13.
Para los biológicos en general (incluyendo los AcMs biosimilares) puede ser difícil evaluar la seguridad a largo plazo únicamente en base a las notificaciones espontáneas de reacciones adversas, por lo que la European Medicines Agency (EMA) puede solicitar la inclusión de los pacientes en registros para promover la captura exhaustiva y consistente de los datos de seguridad1,3. También pueden solicitar estudios adicionales de seguridad postcomercialización. Estos estudios permiten detectar reacciones adversas infrecuentes, que solo se observan cuando se utiliza el fármaco en poblaciones más amplias y durante periodos de tiempo más largos que en los estudios de registro3.
En resumen, la seguridad de los AcMs se monitoriza de forma más exhaustiva que la de la mayoría de fármacos de síntesis química (dada su complejidad), pero no existen requerimientos especiales de farmaco-vigilancia para los biosimilares respecto a los aplicados a los biológicos originales. Los resultados obtenidos desde 2006 avalan la estrategia de la EMA: por el momento no se ha detectado ninguna diferencia relevante en la seguridad de los biosimilares aprobados respecto a sus biológicos originales, y ningún biosimilar se ha retirado por razones de seguridad3.
Extrapolación
Una vez el biosimilar ha demostrado similitud con el biológico original en cuanto a calidad, datos preclínicos y FC/FD, y ha demostrado una eficacia equivalente y seguridad similar en al menos una de sus indicaciones aprobadas, la EMA permite apoyarse en la experiencia ganada con el biológico original para extender la totalidad de la evidencia del biosimilar al resto de indicaciones aprobadas del biológico original. Esto evita repetir ensayos clínicos de fase III de forma innecesaria, con las consiguientes implicaciones éticas y económicas. El único aspecto que no se puede extrapolar directamente es la inmunogenicidad que, como ya se ha comentado, está influenciada por características ajenas al product1,3.
De acuerdo con los requerimientos de la EMA, para permitir la extrapolación se deben cumplir las siguientes condiciones: a) El mecanismo de acción debe estar mediado por la misma molécula diana en ambas indicaciones; (b) el biosimilar debe haber demostrado equivalencia con el biológico original en estudios comparativos realizados en una población suficientemente sensible para detectar diferencias entre ambos, si las hubiera; c) si las indicaciones pertenecen a diferentes áreas terapéuticas, y el mecanismo de acción, posología y/o FC del biosimilar difieren de los del biológico original, pueden ser necesarios estudios adicionales; d) el biosimilar debe haber demostrado un perfil de seguridad comparable con el del biológico original en la indicación evaluada, y e) el biosimilar debe realizar estudios adicionales de inmunogenicidad1,3. Esta estrategia viene avalada por los datos de seguridad y eficacia obtenidos desde que se aprobó el primer biosimilar en la UE en 20063.
Es importante recordar que la extrapolación no es un concepto completamente nuevo; se asemeja al ejercicio de comparabilidad que se aplica de forma rutinaria a los biológicos originales cuando se realizan cambios mayores en su proceso de fabricación. En estos casos, la EMA también se apoya en estudios preclínicos de calidad e in vitro para aplicar la evidencia obtenida con el biológico previo al cambio al biológico obtenido por el nuevo proceso, y no obliga a repetir los ensayos clínicos en cada indicación aprobada3.
Intercambiabilidad, sustitución y switch
Se espera que los biosimilares aprobados para una indicación determinada tengan el mismo efecto clínico que su biológico original. Por tanto, es posible intercambiar un biológico original por su biosimilar (o viceversa), o un biosimilar por otro, mediante switch (prescriptor) o sustitución (farmacéutico). La EMA no proporciona recomendaciones sobre la intercambiabilidad con el biológico original: aunque aconsejan involucrar a los prescriptores en la decisión final, la postura conjunta de la EMA y la Comisión Europea (CE) es que son los Estados miembros los que deben decidir si los biológicos y sus respectivos biosimilares pueden ser intercambiables3.
De acuerdo con la legislación europea, los estudios de switch no son obligatorios para los biosimilares. No obstante, todos los AcMs biosimilares dirigidos a enfermedades inflamatorias14-17 y uno de los disponibles para el tratamiento del cancer17 han incluido uno o, de forma menos frecuente, múltiples switch en sus estudios clínicos de fase III. Una revisión sistemática de la literatura hasta junio de 2017, que consideró también estudios postautorización, identificó 50 estudios con switch de AcMs originales a biosimilares en el área de inflamación. Los autores concluyeron que, en la gran mayoría de estos estudios, no se notificaron diferencias en cuanto a la eficacia, seguridad e inmunogenicidad después del switch. Se ha de destacar que casi todos los estudios identificados en esta revisión implicaron un único switch del biológico original a un biosimilar. Además, los autores no pudieron identificar ningún estudio que notificara un switch entre biosimilares de un mismo biológico original18. Una segunda revisión sistemática de estudios pre y postautorización hasta noviembre de 2017 únicamente identificó dos estudios de switch en indicaciones oncológicas, lo que según los autores podría explicarse por dificultades técnicas y éticas19. Cabe destacar que, al contrario de lo que ocurre con las enfermedades inflamatorias, la naturaleza aguda de muchas indicaciones en oncología implica un uso a corto plazo de AcMs terapéuticos, que dificulta la evaluación de switch. A medida que vaya aumentando el número de AcMs biosimilares disponibles, es previsible que crezca el número de estudios de switch en estos escenarios menos explorados (oncología, switch múltiple). No obstante, el aumento en las opciones de tratamiento también hará difícil abarcar todas las situaciones con las que los prescriptores van a enfrentarse en la práctica clínica. En este sentido, la monitorización postcomercialización, los registros y bases de datos de pacientes, y los estudios de real world evidence pueden proporcionar una información adicional valiosa sobre los diferentes patrones de switch y sus resultados. Esto, junto con los estrictos requerimientos establecidos por la EMA para la aprobación de los AcMs biosimilares, ayudará a reforzar la evidencia obtenida en estudios clínicos sobre la seguridad de intercambiar fármacos con un mismo principio activo biológico20.
Biosimilares de anticuerpos monoclonales actualmente disponibles en la Unión Europea
Obtuvimos el listado de los biosimilares aprobados en la UE (hasta febrero de 2019) en la página web de la EMA (https://www.ema.europa.eu/en/medicines), combinando los filtros “categories=human", “medicine=European public assessment reports (EPAR)", “authorisation status=authorised" y “medicine type=biosimilar". El estatus de aprobación se confirmó mediante el Union Register of Medicinal Products (https://ec.europa.eu/health/documents/community-register/html/index_en.htm). Posteriormente, llevamos a cabo una revisión manual para limitar los resultados a AcMs indicados en las áreas terapéuticas de interés. Para completar la información proporcionada por los EPAR de la EMA y localizar datos relevantes publicados postautorización, llevamos a cabo una búsqueda en PubMed y en los principales congresos de enfermedades inflamatorias/ oncología.
Enfermedades inflamatorias
Los nueve biosimilares de AcMs aprobados en la UE para el tratamiento de las enfermedades inflamatorias (incluyendo reumatología, dermatología y gastroenterología) se concentran en tres moléculas: adalimumab, infliximab y rituximab. Aunque no son anticuerpos, ya que su estructura solo incluye una porción de anticuerpo (región constante de inmunoglobulina G humana), los dos biosimilares aprobados para el inhibidor de factor de necrosis tumoral alfa etanercept se han incluido también debido a su complejidad15,16. La Tabla 1 muestra datos relevantes sobre estos biosimilares.
Tabla 1. Principales características de los biosimilares de anticuerpos monoclonales aprobados en la Unión Europea en el área de las enfermedades inflamatorias hasta febrero de 2019

El farmaco biológico original aparece en negrita. ACR20: mejora del 20% en los criterios establecidos por el American College of Rheumatology; AIJ: artritis idiopatica juvenil; AR: artritis reumatoide; B; biosimilar; DAS28: Disease Activity Score 28; O: farmaco original; PASI: Psoriasis Area and Severity Index; PASI75: mejora ≥ 75% en PASI; Ps: psoriasis; sem.: semana; UE: Union Europea. aTeniendo en cuenta la duracion del tratamiento en el estudio fase III} estudio de extension, con datos disponibles hasta la fecha. bIdacioR (adalimumab) y KromeyaR (adalimumab), ambos de Fresenius Kabi, obtuvieron la opinion positiva del CHMP en enero de 2019. A fecha de 1 de marzo de 2019 no se ha publicado la decision de la CE30,31. cEn pacientes con psoriasis. dEn pacientes con artritis reumatoide. eUnicamente en pacientes adultos. fTambien en pacientes pediatricos. gLos efectos del switch no se evaluaron dentro del estudio de fase III de 52 semanas, sino en otro estudio de 24 semanas llevado a cabo por el titular
Todos los biosimilares de rituximab e infliximab, y la mitad de los de adalimumab (FKB-327, SB5) y etanercept (SB4) han sido aprobados para su uso en enfermedades inflamatorias en base a un único ensayo de fase III en artritis reumatoide, población suficientemente sensible para detectar diferencias, aunque los inmunosupresores que reciben estos pacientes podrían dificultar la evaluación de diferencias en la inmunogenicidad15,17. Para aportar más información, GP2017 y ABP501 (ambos adalimumab) disponen de un ensayo de fase III adicional en pacientes con psoriasis17. Por otra parte, GP2015 (etanercept) se evaluó primero en pacientes con psoriasis16, y posteriormente se han publicado los resultados de un estudio de fase III adicional en artritis reumatoide21,22. En varias de sus evaluaciones de biosimilares, el Comité de Medicamentos de Uso Humano (CHMP, por sus siglas en inglés) ha expresado su preferencia por las variables continuas (por ejemplo, cambio en el Disease Activity Score 28 (DAS28) o el Psoriasis Area and Severity Index (PASI)) respecto a las variables categóricas (por ejemplo, American College of Rheumatology 20 (ACR20) o PASI75), y por las mediciones tempranas de la respuesta (antes de que la curva de respuesta alcance su meseta) respecto a las tardías, por considerarlas más sensibles para detectar potenciales diferencias. Por ello, la mayoría de los estudios han incluido variables continuas de eficacia (principales o secundarias), y las han evaluado en diferentes puntos temporales16,17. De los biosimilares considerados aquí, los que presentan un tratamiento a más largo plazo en pacientes con artritis reumatoide son SB4 (etanercept), y FKB-327 y ABP501 (ambos adalimumab), con > 90 semanas. La duración del tratamiento en los estudios en psoriasis fue muy similar entre los diferentes biosimilares (51-52 semanas)16,17. Como ya se ha comentado, en el programa de desarrollo de todos los biosimilares de AcMs/etanercept aprobados para enfermedades inflamatorias se ha investigado el efecto del switch (en psoriasis, ABP501, GP2017 (ambos adalimumab) y GP2015 (etanercept)), aunque únicamente GP2017 (ambos adalimumab) y GP2015 (etanercept) poseen evidencia sobre switch multiple16,17. En todos los estudios realizados se ha notificado equivalencia en eficacia, seguridad e inmunogenicidad tras uno o múltiples switch. En el periodo postcomercialización, la inmensa mayoría de los estudios de switch se han realizado con CT-P13 (infliximab)18.
Oncología
La CE ha aprobado nueve biosimilares de AcMs para indicaciones en oncología, incluyendo trastuzumab, rituximab y bevacizumab. La Tabla 2 muestra datos relevantes sobre estos biosimilares.
Tabla 2. Principales características de los biosimilares de anticuerpos monoclonales aprobados en la Unión Europea en el área de oncología hasta febrero de 2019

El farmaco biologico original aparece en negrita. CCR: cancer colorrectal; CPNM: cancer de pulmon no microcitico; HER2+: positivo para el receptor 2 del factor de crecimiento epidermico humano; LFA: linfoma folicular avanzado; LLC: leucemia linfocitica cronica; LNH: linfoma no Hodgkin; O-B: del farmaco original al biosimilar; RPC: respuesta patologica completa; RR: riesgo relativo; TRG: tasa de respuesta global; UE: Union Europea. aIncluye linfoma folicular con respuesta al tratamiento de induccion, linfoma folicular de grado III-IV quimiorresistente o refractario/en recaida, y linfoma difuso de celulas B grandes. bSin tratamiento previo o refractaria/en recaida. cAvanzado no resecable, recidivante o metastasico, con predominio de celulas no escamosas. dAvanzado en primera linea, o sensible a platino en primera recaida, o resistente a platino recurrente. eAvanzado y/o metastasico. fPersistente, recurrente o metastasico. gSolo se dispone de datos de switch en reumatologia. hUn segundo estudio de soporte evaluo como variables secundarias la RPC en mama y la TRG en pacientes con cancer de mama HER2+ operable. iMientras MVASI y Zirabev unicamente estan indicados en combinacion con paclitaxel, Avastin esta indicado ademas en combinacion con capecitabina en pacientes en los que no se considera apropiado el tratamiento con taxanos o antraciclinas. jA diferencia de Avastin y MVASI, Zirabev unicamente esta indicado para el tratamiento de CPNM sin mutaciones activadoras del receptor del factor de crecimiento epidermico (EGFR).
CT-P10 y GP2013, biosimilares de rituximab, se han evaluado en pacientes con linfoma folicular avanzado, por ser la indicación aprobada de rituximab más común en oncología y suficientemente sensible para detectar potenciales diferencias entre el biosimilar y el biológico original. La variable principal escogida en ambos casos ha sido la tasa de respuesta global (TRG), relevante en esta indicación según el CHMP17.
Todos los estudios de fase III de los biosimilares de trastuzumab se han realizado en pacientes con cáncer de mama, porque el mecanismo de acción descrito en cáncer de mama precoz y metastásico HER2+, y en cáncer gástrico metastásico HER2+, es similar. La población de pacientes con cáncer de mama precoz HER2+ en neoadyuvancia y adyuvancia, incluida en los estudios confirmatorios de SB3, ABP980 y CT-P6 se considera más sensible para evaluar posibles diferencias que la población con cáncer metastásico incluida en el estudio de fase III de MYL-1401O y en el principal estudio de fase III de PF-05280014, aunque este último también dispone de un estudio de apoyo en neoadyuvancia. El CHMP, no obstante, ha validado ambas aproximaciones. De la misma manera, aunque el CHMP ha avalado la sensibilidad de la variable principal escogida para CT-P6 y ABP980 (respuesta patológica completa (RPC) total, ausencia de cáncer invasivo tanto en la mama como en los ganglios axilares), también ha considerado aceptable la variable principal escogida en el estudio de SB3 (RPC únicamente en mama)17. Los efectos del switch solo se han evaluado para ABP98017,19, y los datos sugieren que el cambio del biológico original al biosimilar no afectó a la eficacia, seguridad o inmunogenicidad.
La patología escogida para los estudios de fase III de los biosimilares de bevacizumab ABP215 y PF-06439535 (cáncer de pulmón no microcítico) también ha sido avalado por el CHMP como suficientemente sensible, y la variable principal evaluada (TRG) es considerada la más sensible para poder detectar diferencias entre el biológico original y sus biosimilares17.
Situación actual y perspectivas futuras de los biosimilares en España
La contribución de los biosimilares a la eficiencia del sistema sanitario es un hecho reconocido por varias sociedades médicas y farmacéuticas españolas6,32-24. Por un lado, los ahorros se pueden atribuir directamente a la adquisición de biosimilares en lugar de biológicos originales. El procedimiento de aprobación abreviado de los biosimilares elimina partes del dosier de registro que son obligatorias para los biológicos originales (por ejemplo, estudios de fase II, buena parte de los estudios de fase III). Como resultado, el precio de lanzamiento estimado para los biosimilares en España es como media un 30% más bajo que el de los biológicos originales35. Por otro lado, los biosimilares pueden inducir ahorros indirectos para el sistema sanitario, impulsados por los requerimientos legales y la competencia. En España, en concreto, cuando se comercializa el primer biosimilar de un biológico original (siempre a un precio reducido respecto al producto de referencia), el precio del biológico original debe reducirse hasta, como mínimo, igualar el precio del biosimilar36. Esto, en teoría, difumina la ventaja de precio que representaría un incentivo para el uso del biosimilar. Sin embargo, se debe tener en cuenta que los fabricantes pueden ofrecer descuentos adicionales en negociaciones posteriores o concursos públicos. Además, a medida que aumentan los biosimilares comercializados para un biológico original concreto, aumenta la competencia, produciendo una bajada de precios aún mayor. Una manera frecuente de aumentar esta competencia son, de nuevo, los concursos públicos, especialmente cuando los contratos son de corta duración y/o se otorgan a varios proveedores simultáneamente37.
La contribución de cada uno de estos escenarios al ahorro de costes sanitarios depende del grado de penetración de los biosimilares en el sistema. Como muestra de esto, un análisis retrospectivo de datos españoles estimó el ahorro derivado de la aparición de los biosimilares en 479 millones de euros para el periodo 2009-2016. Más de la mitad (65%) de este ahorro se produjo entre 2015-2016, principalmente por la bajada de precio de los biológicos originales infliximab e insulina glargina impulsada por la aparición de biosimilares, y no por la adquisición directa de éstos. El mismo análisis estimó un ahorro de 1.965 millones de euros para el periodo 2017-2020, por la aparición de biosimilares para el tratamiento de patologías muy prevalentes y/o que actualmente se tratan con biológicos originales de precio Elevado35. El ahorro derivado directa o indirectamente de los biosimilares libera recursos que pueden invertirse en nuevos tratamientos o tecnologías sanitarias originales. La adopción de estas terapias, además de proporcionar un beneficio inmediato al paciente, supone un impulso a la innovación en la industria farmacéutica, que deriva a su vez en beneficios adicionales para los pacientes a largo plazo. El efecto de los biosimilares en la innovación se ve reforzado además por la aparición de nuevos dispositivos de administración asociados a algunos biosimilares, y la realización de estudios clínicos o de real world evidence adicionales. Esto último es especialmente relevante, ya que la toma de decisiones por parte de los prescriptores depende en gran medida de la evidencia científica. En este sentido, se debe destacar que el proceso de aprobación abreviado de los similares (justificado por la totalidad de la evidencia disponible) no ha ido en detrimento de su eficacia y seguridad, como muestran los datos recogidos por la EMA hasta el momento. Como se ha comentado anteriormente, los estudios de fase III de los biosimilares han incluido poblaciones y variables que cumplen los requerimientos del CHMP y, en algunos casos, las compañías han realizado estudios de apoyo en indicaciones reclamadas históricamente por las sociedades médicas, como la psoriasis32. Asimismo, los programas de desarrollo han incluido características en principio no requeridas por la EMA para demostrar biosimilitud, pero que pueden ser de interés para los prescriptores, como son los estudios de switch. Es de prever que, si se dirigen esfuerzos a comunicar esta evidencia a los prescriptores de forma efectiva, se ayude a vencer las reticencias que todavía puedan quedar respecto a los biosimilares, lo cual ayudaría a su vez a aumentar su penetración en el sistema sanitario sin tener que imponer necesariamente cuotas de prescripción.
Otro aspecto que puede ayudar a aumentar el uso de los biosimilares, por su influencia en los prescriptores, es el posicionamiento favorable (o como mínimo neutro) de las sociedades médicas y la mención expresa a los biosimilares en las guías de tratamiento. En el primer punto se ha avan-zado mucho, y en la actualidad varias sociedades científicas españolas avalan el procedimiento de generación de evidencia establecido por la EMA para los biosimilares. Como ejemplo, la Sociedad Española de Farmacia Hospitalaria (SEFH), la Sociedad Española de Patología Digestiva (SEPD), la SEOM y la Academia Española de Dermatología y Venereología (AEDV) aceptan actualmente la extrapolación, siempre que se cumplan los requerimientos de la EMA6,32,34,38. Respecto a la mención de los biosimilares en las guías clínicas, sigue siendo una asignatura pendiente, también se han hecho algunos avances. Un ejemplo son las recomendaciones de la Sociedad Española de Reumatología sobre el uso de terapias biológicas en pacientes con espondiloartritis axial39.
La confianza en el perfil de seguridad de los biosimilares es otro aspecto clave. Ya se dispone de información de seguridad a largo plazo proveniente de ensayos clínicos, y todos los biosimilares tienen un plan de gestión de riesgo y están sujetos a los mismos requerimientos de farmacovigilancia postcomercialización que los biológicos originales. Además, a medida que transcurra el periodo postcomercialización, se generarán datos en práctica clínica en poblaciones más amplias y diversas que las incluidas en los ensayos clínicos. La trazabilidad jugará un papel cada vez más importante a medida que aparezcan nuevos biosimilares y aumenten las opciones terapéuticas para un mismo principio activo. El identificador único impreso en todos los envases de medicamentos de prescripción a partir de febrero de 201940 facilita la trazabilidad de estos fármacos y ayuda a atribuir los posibles acontecimientos adversos que puedan ocurrir a fármacos concretos, dilucidando el perfil de seguridad de unos biosimilares respecto a otros y respecto a los biológicos originales.
A nivel administrativo, el acceso de los biosimilares se podría agilizar facilitando su inclusión en las guías farmacoterapéuticas de los hospitales. Actualmente, esta inclusión se suele consensuar entre el servicio médico, el servicio de farmacia hospitalaria, y la dirección médica y económica, o bien se decide en la Comisión de Farmacia y Terapéutica. Por otro lado, en los casos en que se convoquen concursos públicos, se deberían otorgar contratos cortos a múltiples ganadores para promover la competencia y, por tanto, la disponibilidad de biosimilares a largo plazo. La concesión de los contratos no debería obedecer únicamente a aspectos económicos, sino también de calidad, incluyendo entre otros la evidencia científica aportada, la disponibilidad de programas de soporte a pacientes, la calidad del material de acondicionamiento, la información incluida en el etiquetado o el dispositivo de administración. Esto contribuiría a aumentar la confianza, compromiso y preferencia de los pacientes y de los profesionales sanitarios por los AcMs biosimilares.
Si consideramos el switch y la sustitución, en España la orden SCO/2874/2007 impide la sustitución automática cuando no hay un consenso previo con el prescriptor41. En consecuencia, los servicios de farmacia solo pueden sustituir biológicos originales por sus biosimilares si lo han consensuado previamente con los prescriptores en la Comisión de Farmacia y Terapéutica, y siempre deben informar al prescriptor del fármaco utilizado en cada caso. Muchos prescriptores prefieren seguir decidiendo qué biosimilar o fármaco original se debe utilizar en cada caso, y persisten prejuicios contra el uso de biosimilares que no están justificados a la luz de la evidencia disponible. Los prescriptores favorables al switch, mientras tanto, pueden encontrar difícil comparar las diferentes ofertas de los biosimilares en todas sus posibles dimensiones. Asimismo, puede ser especialmente difícil que diferentes servicios de un mismo centro lleguen a un consenso sobre la intercambiabilidad de un determinado biosimilar, especialmente cuando la disponibilidad de estudios sobre switch varía ostensiblemente entre las diferentes áreas terapéuticas.
A nivel nacional, países como Portugal, Francia y Reino Unido someten a los biosimilares aprobados por la CE a una evaluación económica adicional de coste/beneficio antes de decidir el reembolso. En España no se requiere esta evaluación para la negociación de precio y reembolso, por lo que no se retrasa tanto la comercialización. No obstante, España no ha establecido todavía procedimientos de negociación abreviados, como se ha hecho en países como Alemania e Italia35. La adopción de medidas para fomentar el uso de biosimilares (formación, estímulos a la prescripción, fijación de objetivos de uso/penetración) también es competencia de los Estados miembros de la UE. En España se han fijado cuotas en algunas regiones (Madrid, Cataluña)35, pero esta estrategia puede ser contraproducente en cuanto implica coartar la libertad de prescripción por razones económicas. Por tanto, en caso de implantarse, debería hacerse de manera que se preserve la capacidad de decisión del prescriptor, fomentando por ejemplo el inicio del tratamiento con biosimilares en pacientes naïve, en lugar de obligar al switch en pacientes con tratamientos ya establecidos. Asimismo, la aceptación de las cuotas de prescripción podría mejorar si el ahorro conseguido con el uso de biosimilares se reinvierte en la atención sanitaria, y la medida se comunica adecuadamente. Adicionalmente, a nivel nacional, se necesitan normativas específicas sobre intercambiabilidad. La adopción de estas medidas agilizaría la entrada de nuevos biosimilares en el sistema sanitario español, aumentando las opciones para los prescriptores y pacientes. Por otro lado, reduciría la incertidumbre entre las compañías implicadas en el desarrollo de biosimilares, estimulando la innovación35.
Finalmente, se deben tener en cuenta también los factores relacionados con los pacientes. En particular, los pacientes que desarrollan una actitud negativa hacia el fármaco que reciben pueden experimentar un empeoramiento subjetivo de sus síntomas, conocido como “efecto nocebo"42. Este efecto ha sido documentado en estudios observacionales con AcMs biosimilares43, y puede ser especialmente relevante en el caso de pacientes que se autoadministran el fármaco, ya que están más familiarizados con su medicación habitual. En este contexto, el papel de los prescriptores y farmacéuticos es especialmente relevante, pues pueden transmitir a los pacientes su confianza en los biosimilares. Al mismo tiempo, pueden proporcionarles información suficiente y fácil de entender, e implicarles en las decisiones terapéuticas, de forma que cualquier switch se acuerde con el paciente. Como esto se puede ver dificultado por la carga de trabajo de los profesionales sanitarios, los pacientes deberían tener acceso a recursos de información fiables, fáciles de localizar y entender. Un ejemplo de esto es el documento sobre biosimilares dirigido a los pacientes que está disponible en la página web de la CE44.
En conclusión, la regulación europea para la aprobación de los biosimilares de AcMs asegura que estos productos sean altamente similares a sus biológicos originales en términos de calidad, eficacia y seguridad. Es de prever que la penetración de los biosimilares en el sistema sanitario se incremente a medida que los prescriptores sean más conscientes de esta opción, y aumente su conocimiento sobre la totalidad de la evidencia que justifica aspectos como la extrapolación y el switch. No obstante, se requieren medidas nacionales/institucionales adicionales que agilicen el acceso a los biosimilares y apoyen la innovación. La educación e implicación de los pacientes en el proceso de decisión serán puntos clave para aumentar la aceptación de los biosimilares y contrarrestar el efecto nocebo.
Agradecimientos
Los autores agradecen la colaboración de Beatriz del Val Romero de TFS S.L. en la redacción del manuscrito.
REFERENCIAS
1. European Medicines Agency. Guideline on biosimilar biological medicinal products (Internet). 2014 (accessed 10/18/2018). Available at: https://www.ema.europa.eu/documents/scientific-guideline/guideline-similar-biological-medicinalproducts-rev1_en.pdf [ Links ]
2. Markus R, Liu J, Ramchandani M, Landa D, Born T, Kaur P. Developing the totality of evidence for biosimilars: regulatory considerations and building confidence for the healthcare community. BioDrugs. 2017(31):175-87. [ Links ]
3. European Medicines Agency, European Commission. Biosimilars in the EU. Information guide for healthcare professionals (Internet). 2017 (accessed 10/18/2018). Available at: https://www.ema.europa.eu/documents/leaflet/biosimilars-euinformation-guide-healthcare-professionals_en.pdf [ Links ]
4. O'Callaghan J, Barry SP, Bermingham M, Morris JM, Griffin BT. Regulation of biosimilar medicines and current perspectives on interchangeability and policy. Eur J Clin Pharmacol. 2018;75(1):1-11. [ Links ]
5. European Medicines Agency. Guidelines on similar biological products containing monoclonal antibodies - nonclinical and clinical issues (Internet). 2012 (accessed 10/18/2018). Available at: https://www.ema.europa.eu/documents/scientificguideline/guideline-similar-biological-medicinal-products-containing-monoclonalantibodies-non-clinical_en.pdf [ Links ]
6. Sociedad Espanola de Oncologia Medica. Posicionamiento SEOM sobre los anticuerpos biosimilares (Internet). 2018 (accessed 10/29/2018). Available at: https://seom.org/seomcms/images/stories/recursos/Posicionamiento_sobre_biosimilares_mayo_2018.pdf [ Links ]
7. Schellekens H. Immunologic mechanisms of EPO-associated pure red cell aplasia. Best Pract Res Clin Haematol. 2005;18(3):473-80. [ Links ]
8. European Medicines Agency. Guidelines on immunogenicity assessment of monoclonal antibodies intended for in vivo clinical use (Internet). 2012 (accessed 10/18/2018). Available at: https://www.ema.europa.eu/documents/scientificguideline/guideline-immunogenicity-assessment-monoclonal-antibodies-intendedvivo-clinical-use_en.pdf [ Links ]
9. Klein AV, Wang J, Feagan BG, Omoto M. Biosimilars: state of clinical and regulatory science. J Pharm Pharm Sci. 2017;20(1):332-48. [ Links ]
10. Agencia Espanola de Medicamentos y Productos Sanitarios. Medicamentos sujetos a seguimiento adicional (Internet). 2016 (accessed 10/25/2018). Available at: https://www.aemps.gob.es/vigilancia/medicamentosUsoHumano/seguimiento_adicional.htm [ Links ]
11. European Comission. Directive 2001/83/EC of the European Parliament and of the Council of 6 November 2001 on the Community code relating to medicinal products for human use (Internet). 2001 (accessed 10/23/2018). Available at: https://ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/dir_2001_83_consol_2012/dir_2001_83_cons_2012_en.pdf [ Links ]
12. Ministerio de Sanidad, Servicios Sociales e Igualdad. Real Decreto 577/2013, de 26 de julio, por el que se regula la farmacovigilancia de medicamentos de uso humano (Internet). 2013 (accessed 10/29/2018). Available at: https://www.boe.es/buscar/pdf/2013/BOE-A-2013-8191-consolidado.pdf [ Links ]
13. Klein K, Stolk P. Challenges and opportunities for the traceability of (biological) medicinal products. Drug Saf. 2018;41(10):911-8. [ Links ]
14. Emery P, Vencovsky J, Sylwestrzak A, Leszczyński P, Porawska W, Stasiuk B, et al. Long-term efficacy and safety in patients with rheumatoid arthritis continuing on SB4 or switching from reference etanercept to SB4. Ann Rheum Dis. 2017;76(12):1986-91. [ Links ]
15. European Medicines Agency. Medicines (Internet). 2015 (accessed 02/28/2019). Available at: https://www.ema.europa.eu/en/medicines [ Links ]
16. European Medicines Agency. Medicines (Internet). 2017 (accessed 02/28/2019). Available at: https://www.ema.europa.eu/en/medicines [ Links ]
17. European Medicines Agency. Medicines (Internet). 2018 (accessed 02/28/2019). Available at: https://www.ema.europa.eu/en/medicines [ Links ]
18. Cohen HP, Blauvelt A, Rifkin RM, Danese S, Gokhale SB, Woollett G. Switching reference medicines to biosimilars: a systematic literature review of clinical outcomes. Drugs. 2018;78(4):463-78. [ Links ]
19. Declerck P, Bakalos G, Zintzaras E, Barton B, Schreitmuller T. Monoclonal antibody biosimilars in oncology: critical appraisal of available data on switching. Clin Ther. 2018;40(5):798-809. [ Links ]
20. Trifiro G, Marciano I, Ingrasciotta Y. Interchangeability of biosimilar and biological reference product: updated regulatory positions and pre-and post-marketing evidence. Expert Opin Biol Ther. 2018;18(3):309-15. [ Links ]
21. Matucci-Cerinic M, Allanore Y, Kavanaugh A, Buch MH, Schulze-Koops H, Kucharz EJ, et al. Efficacy, safety and immunogenicity of GP2015, an etanercept biosimilar, compared with the reference etanercept in patients with moderate-to-severe rheumatoid arthritis: 24-week results from the comparative phase III, randomised, double-blind EQUIRA study. RMD Open. 2018;4(2):e000757. [ Links ]
22. Kavanaugh A, Matucci-Cerinic M, Schulze-Koops H, Buch M, Allanore Y, Kucharz EJ, et al. Phase 3 Equira 48 week study results demonstrated no impact on efficacy and safety when patients with moderate-to-severe rheumatoid arthritis were switched between reference etanercept (ETN) and GP2015, an etanercept biosimilar. Presentado en: ACR/ARHP Annual Meeting; 2018; Chicago (USA). Abstract 2550. [ Links ]
23. European Medicines Agency. Medicines (Internet). 2019 (accessed 02/28/2019). Available at: https://www.ema.europa.eu/en/medicines [ Links ]
24. Cohen S, Pablos JL, Pavelka K, Muller GA, Matsumoto A, Kivitz A, et al. An openlabel extension study to demonstrate long-term safety and efficacy of ABP 501 in patients with rheumatoid arthritis. Arthritis Res Ther. 2019;21(1):84. [ Links ]
25. Wiland P, Jeka S, Dokoupilova E, Miranda-Limon JM, Jauch-Lembach J, Thakur A, et al. A randomized, double-blind, parallel-group, multicenter study to compare the efficacy, safety and immunogenicity of a proposed adalimumab biosimilar (GP2017) with reference adalimumab in patients with moderate-to-severe active rheumatoid arthritis. Rheumatol. 2018;70(Suppl 10):Abstract 1936. [ Links ]
26. Yoo DH, Prodanovic N, Jaworski J, Miranda P, Ramiterre E, Lanzon A, et al. Efficacy and safety of CT-P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT-P13 and continuing CT-P13 in the PLANETRA extension study. Ann Rheum Dis. 2017;76(2):355-63. [ Links ]
27. Smolen JS, Choe JY, Prodanovic N, Niebrzydowski J, Staykov I, Dokoupilova E, et al. Safety, immunogenicity and efficacy after switching from reference infliximab to biosimilar SB2 compared with continuing reference infliximab and SB2 in patients with rheumatoid arthritis: results of a randomised, double-blind, phase III transition study. Ann Rheum Dis. 2018;77(2):234-40. [ Links ]
28. Cohen S, Kivitz AJ, Tee M, Cronenberger C, Zhang M, Hackley S, et al. A randomized, double-blind phase III study comparing the efficacy, safety and immunogenicity of PF-06438179/GP1111 (IxifiTM), an infliximab biosimilar, and infliximab reference product (RemicadeR) in patients with moderate to severe active RA: results from week 54 to week 78. Arthritis Rheumatol. 2018;70(Suppl 10):Abstract 2521. [ Links ]
29. Tony HP, Schulze-Koops H, Kruger K, Cohen SB, Kivitz AJ, Jeka S, et al. Comparison of switching from the originator rituximab to the biosimilar rituximab GP2013 or re‑treatment with the originator rituximab in patients with active rheumatoid arthritis: safety and immunogenicity results from a multicenter, randomized, double-blind study. Arthritis Rheumatol. 2017;69(Suppl 10):Abstract 2795. [ Links ]
30. European Medicines Agency. Idacio summary of opinion (Internet). 2019 (accessed 01/03/2019). Available at: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-idacio_en.pdf [ Links ]
31. European Medicines Agency. Kromeya summary of opinion (Internet). 2019 (accessed 01/03/2019). Available at: https://www.ema.europa.eu/en/documents/smop-initial/chmp-summary-positive-opinion-kromeya_en.pdf [ Links ]
32. Carretero Hernandez G, Puig L, en representacion del Grupo de Psoriasis de la AEDV. The use of biosimilar drugs in psoriasis: a position paper. Actas Dermosifiliogr. 2015;106(4):249-51. [ Links ]
33. Sociedad Espanola de Reumatologia. Documento de posicionamiento de la Sociedad Espanola de Reumatologia sobre farmacos biosimilares. Actualizacion 2018 (Internet). 2018 (accessed 10/29/2018). Available at: https://www.ser.es/wp-content/uploads/2018/01/POSICIONAMIENTO-SER-2018.pdf [ Links ]
34. Martinez-Lopez de Castro N, Matilla-Fernandez MB, Fraga-Fuentes MD, Mangues-Bafalluy I, Asensi-Diez R, Cajaraville-Ordonana G. Spanish Society of Hospital Pharmacy position paper on biosimilar medicines. Farm Hosp. 2018;42(4):180-3. [ Links ]
35. Fundacion Gaspar Casal. Libro blanco de los medicamentos biosimilares en Espana: Innovacion y sostenibilidad (Internet). 2017 (accessed 10/26/2018). Available at:http://fundaciongasparcasal.org/publicaciones/Libro-Blanco-de-los-Medicamentos-Biosimilares-en-Espana-Innovacion-y-Sostenibilidad.pdf [ Links ]
36. Ministerio de Sanidad, Servicios Sociales e Igualdad. Real Decreto 177/2014, de 21 de marzo, por el que se regula el sistema de precios de referencia y de agrupaciones homogeneas de medicamentos en el Sistema Nacional de Salud, y determinados sistemas de informacion en materia de financiacion y precios de los medicamentos y productos sanitarios (Internet). 2014 (accessed 10/25/2018). Available at: https://www.boe.es/diario_boe/txt.php?id=BOE-A-2014-3189 [ Links ]
37. IQVIA Institute for Human Data Science. Advancing biosimilar sustainability in Europe. A multi-stakeholder assessment (Internet). 2018 (accessed 10/25/2018). Available at: https://www.iqvia.com/-/media/iqvia/pdfs/institute-reports/advancingbiosimilar-sustainability-in-europe.pdf?=1540474083504 [ Links ]
38. Arguelles Arias F, Hinojosa Del Val J, Vera Mendoza I. Update of the SEPD position statement on the use of biosimilars for inflammatory bowel disease. Rev Esp Enferm Dig. 2018;110(6):407. [ Links ]
39. Sociedad Espanola de Reumatologia. Recomendaciones de la Sociedad Española de Reumatologia (SER) sobre el uso de terapias biologicas en espondiloartritis axial (Internet). 2017 (accessed 01/03/2019). Available at: https://www.ser.es/wpcontent/uploads/2017/04/RECOMENDACIONES-Terapias-Biolo%C3%B3gicasen-la-EspAax.pdf [ Links ]
40. European Comission. Reglamento delegado (UE) 2016/161 de la Comision de 2 de octubre de 2015 que completa la Directiva 2001/83/CE del Parlamento Europeo y del Consejo estableciendo disposiciones detalladas relativas a los dispositivos de seguridad que figuran en el envase de los medicamentos de uso humano (Internet). 2016 (accessed 10/25/2018). Available at: https://ec.europa.eu/health//sites/health/files/files/eudralex/vol-1/reg_2016_161/reg_2016_161_es.pdf [ Links ]
41. Ministerio de Sanidad y Consumo. Orden SCO/2874/2007, de 28 de septiembre, por la que se establecen los medicamentos que constituyen excepción a la posible sustitucion por el farmaceutico con arreglo al articulo 86.4 de la Ley 29/2006, de 26 de julio, de garantias y uso racional de los medicamentos y productos sanitarios (Internet). 2007 (accessed 10/24/2018). Available at: https://www.boe.es/buscar/pdf/2007/BOE-A-2007-17420-consolidado.pdf [ Links ]
42. Kristensen LE, Alten R, Puig L, Philipp S, Kvien TK, Mangues MA, et al. Non-pharmacological effects in switching medication: the nocebo effect in switching from originator to biosimilar agent. BioDrugs. 2018;32(5):397-404. [ Links ]
43. Bakalos G, Zintzaras E. Drug discontinuation in studies including a switch from an originator to a biosimilar monoclonal antibody: a systematic literature review. Clin Ther. 2018;41(1):155-73. [ Links ]
44. European Comission. Que necesito saber sobre los medicamentos biosimilares. Información para pacientes (Internet). 2016 (accessed 10/22/2018). Available at: https://ec.europa.eu/growth/content/information-patients-what-i-need-know-aboutbiosimilar-medicines-0_en 2016 [ Links ]
Recibido: 20 de Mayo de 2019; Aprobado: 28 de Febrero de 2020
 This is an open-access article distributed under the terms of the Creative Commons Attribution License
This is an open-access article distributed under the terms of the Creative Commons Attribution License
