










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
La fibrosis quística (FQ) es una enfermedad causada por la mutación de un gen que codifica el canal clorhídrico transmembrana denominado regulador de conductancia transmembrana de la fibrosis quística (CFTR), que regula el transporte de aniones y aclaramiento mucociliar en las vías aéreas1. En la Figura 1 se recoge un esquema que ilustra el transporte iónico a través de canales en una célula normal y una célula afectada de la enfermedad. El fallo funcional en el CFTR resulta en la retención de moco e infección crónica y, subsiguientemente, en la inflamación de las vías aéreas. Este efecto resulta seriamente perjudicial para los pulmones.
La disfunción del gen CFTR afecta principalmente a las células epiteliales, aunque existen evidencias de que las células inmunes ejercen también un papel importante2.

Figura 1. Representación esquemática de los canales iónicos de sodio y cloruro en la fibrosis quística, comparados con los mismos en células normales.
La FQ afecta a varios sistemas corporales, y la morbi-mortalidad está causada principalmente por bronquiectasias, pequeñas obstrucciones de las vías aéreas y discapacidad respiratoria progresiva3. Además, es frecuente la aparición de problemas en otros órganos como el páncreas (malabsorción), hígado (cirrosis hepática), glándulas sudoríparas (golpe de calor) y otras complicaciones, como infertilidad.
Por todo ello, el diseño y desarrollo de sistemas que mejoren el aclaramiento de moco en los pulmones y la posterior infección que se origina, junto con la mejora de la insuficiencia pancreática y la desnutrición, por parte de equipos multidisciplinares, daría lugar a importantes avances en la calidad de vida y en los resultados clínicos de los pacientes con FQ, que ahora tienen una esperanza de vida no superior a 40 años4.
A pesar de que se siguen empleando los tratamientos convencionales por vía oral e intravenosa, en los últimos años se están implementando terapias alternativas, tanto aprovechando la vía pulmonar como ruta de acceso directo a la zona afectada, como atacando al defecto de base en la FQ. Estos nuevos tratamientos suelen ser bastante efectivos ya que mejoran la función pulmonar, reduciendo asimismo las exacerbaciones a nivel de este órgano.
Sin embargo, el uso de la vía pulmonar presenta limitaciones que giran, sobre todo, en torno a su modo de administración. El rápido aclaramiento de los fármacos da lugar a bajos niveles tisulares y por ello se debe incrementar la frecuencia de administración. Asimismo, es frecuente la aparición de efectos adversos. Con el fin de soslayar estos inconvenientes, la utilización de nanosistemas se postula como estrategia interesante para el tratamiento de esta enfermedad5.
En base a todo ello, el objetivo del presente trabajo se centra en revisar y analizar cómo la nanotecnología se ha ido implantando en la investigación del desarrollo de formulaciones adecuadas para tratar la FQ. De todos los nanotransportadores existentes, la revisión se centrará en los de tipo lipídico, y concretamente, en los liposomas, por su biocompatibilidad y similitud con el surfactante pulmonar.
MÉTODOS
Se ha realizado una revisión no sistemática de documentos científicos dedicados a la evolución de los tratamientos de la FQ, destacando la importancia de la vía pulmonar y centrándonos en la aportación de la nanotecnología.
Para ello, se realizó una búsqueda bibliográfica en diferentes bases de datos, como Science Direct y PubMed, principalmente en los últimos 10 años, aunque en algunos casos, se tuvo que acudir a referencias anteriores. Se inició la búsqueda con palabras clave combinadas, como fibrosis quística (cystic fibrosis) y administración pulmonar de fármacos (pulmonary drug delivery), fibrosis quística (cystic fibrosis) y sistemas nanoparticulares (nanoparticle, nanotechnology, nanocarrier). De la totalidad de los artículos encontrados, se seleccionaron aquellos de interés en el tema de la revisión como criterio de exclusión o inclusión. La siguiente etapa consistió en acotar el campo de los sistemas nanoparticulares y realizar la búsqueda particular de ellos; en concreto, liposomas (liposomes) y fibrosis quística (cystic fibrosis).
RESULTADOS
Esta parte del artículo se estructuró en tres bloques principales: fibrosis quística, importancia de la vía pulmonar para la administración de fármacos y sistemas nanoparticulares para la administración de estos.
FIBROSIS QUÍSTICA
Los notables progresos que se han conseguido en los últimos años en torno al tratamiento de esta enfermedad se deben fundamentalmente a la mejora del aclaramiento mucociliar de las vías aéreas y al control de la infección pulmonar. De esta forma, se ha conseguido que la FQ deje de ser una enfermedad predominante en niños para convertirse en una enfermedad de adultos6; de hecho, el número de adultos con FQ continuará aumentando, produciéndose casi todas las muertes en este grupo de población7. Tratando cifras, en los últimos cinco años, en los países con sistemas sanitarios desarrollados, ha habido más adultos que niños con FQ. Se prevé que en los países europeos desarrollados, el número de adultos con FQ aumente un 70% en 20258, mientras que en los países de Europa con sistemas sanitarios menos desarrollados, la esperanza de vida puede quedarse en la segunda década debido a una falta de acceso a los tratamientos9.
La sustancial mejora de la esperanza de vida en los países desarrollados ha sido fruto de la existencia de centros bien organizados y multidisciplinares, así como del empleo de fármacos efectivos para tratar la infección y mejorar el aclaramiento mucociliar10.
La FQ se identifica normalmente mediante el cribado a los recién nacidos o durante los primeros años de vida11. Las personas que son diagnosticadas después de los 20 años de edad tienen normalmente una mutación asociada con la función residual de CFTR, como por ejemplo Arg117His, también conocida como R117H12.
Estos pacientes pueden tener síntomas de taponamiento respiratorio en la infancia, pero desarrollan bronquiectasias, pancreatitis o presentan infertilidad más adelante. El diagnóstico se realiza mediante el test del sudor y el análisis del ADN13. Los individuos con un diagnóstico tardío tienen una buena supervivencia, lo que refleja la elevada prevalencia de mutaciones asociadas con la función residual y con un fenotipo menos grave13.
El tratamiento de la enfermedad abarca el uso de fármacos para atender tanto las causas como los síntomas. Así, las dianas del tratamiento sintomatológico incluyen:
Aparato respiratorio
Tratamiento antibiótico oral
El manejo de la enfermedad respiratoria siempre ha sido fundamental en el tratamiento de la enfermedad ya que el fallo respiratorio provocado por la infección es la segunda causa de muerte por esta enfermedad.
Las fluoroquinolonas han demostrado eficacia como tratamiento antibiótico oral. Entre ellas destaca ciprofloxacino y levofloxacino. Además, otros fármacos como tobramicina y amikacina pueden ser efectivos en la prevención de las exacerbaciones pulmonares asociadas a las infecciones del tracto respiratorio, especialmente en aquellas causadas por Pseudomonas aeruginosa, ya que esta bacteria es la que más comúnmente causa infecciones pulmonares en enfermos de FQ.
Los antibióticos empleados para el tratamiento de exacerbaciones pulmonares se suelen administrar por vía intravenosa, mientras que para tratamientos prolongados se emplean inhaladores o medicamentos por vía oral14.
Broncodilatadores
Se emplean los ß2agonistas, que tienen un efecto directo en la relajación de la musculatura lisa y aumentan la frecuencia del barrido mucociliar (que consiste en la secreción continuada de moco, el cual mantiene la hidratación de las vías aéreas). Sin embargo, el aumento de la viscosidad del esputo puede hacer disminuir este beneficio por lo que la respuesta a estos fármacos frecuentemente es muy variable. Dentro de este grupo, los fármacos más usados son salbutamol, y fenoterol o salbutamol en mezcla con bromuro de ipratropio. Se ha demostrado que estas moléculas deberían usarse cuando se hace ejercicio y para pacientes que presenten sibilancias.
También se emplea teofilina, base xántica con efecto broncodilatador, estimulando el centro respiratorio. Se trata de un inhibidor de la fosfodiesterasa VI, enzima que degrada el AMPc intracelular. El incremento de los niveles de AMPc induce la relajación del músculo liso bronquial, inhibiendo asimismo mediadores químicos a nivel mastocitario15. Sus dos acciones farmacológicas son la relajación del músculo liso bronquial (tanto a nivel central como periférico) y su acción antiinflamatoria, ya que inhibe la liberación de los mediadores químicos mastocitarios que participan en los mecanismos inflamatorios a nivel bronquial. Actualmente, se administra tanto por vía oral como por vía intravenosa. La administración por vía pulmonar supondría un aumento del grado de cumplimiento por parte del paciente16.
Corticoides orales e inhalados
Existen algunas sugerencias que relacionan el defecto básico en la FQ y el inicio de la inflamación, con la desregulación de la producción de citoquinas, lo que favorecería un proceso inflamatorio persistente. Los corticoides actúan disminuyendo la inflamación producida. Uno de los que se emplea por vía oral normalmente es prednisona, 1 mg/kg/día durante 5 a 7 días. Por vía inhalatoria, se emplean en aquellos pacientes que demuestren hiperreactividad bronquial. Los más utilizados son beclometasona, budesonida y fluticasona16.
Aparato gastrointestinal
El tratamiento se basa principalmente en prevenir o tratar las obstrucciones intestinales. Para ello, se emplean sueros para rehidratar o laxantes osmóticos, para las obstrucciones incompletas; o enemas hiperosmolares, para las obstrucciones completas. Uno de los problemas emergentes de la FQ son las complicaciones gastrointestinales, como cáncer de colon y pólipos intestinales. La prevención de éstos es primordial para asegurar la supervivencia del paciente.
La insuficiencia pancreática se trata con terapia pancreática enzimática de reemplazo, que contiene una combinación múltiple de proteasas, amilasas y lipasas17.
Nutrición y electrolitos
Se recomienda una nutrición adecuada y la prevención de la deshidratación. Se aconseja una dieta hipercalórica rica en grasas saludables con suplementos de vitaminas A, D, E y K; y de elementos, como fluoruro y cinc. La suplementación con cloruro sódico depende de la edad del paciente y de sus condiciones ambientales18.
Tratamiento actual y del futuro
Las terapias actuales y las que tendrán éxito en un futuro próximo se basan principalmente en corregir las anormalidades estructurales y funcionales del gen CFTR. Entre los compuestos que se utilizan destacan los moduladores del CFTR, que son capaces de corregir el defecto básico de la FQ, el que ocurre en la proteína. El mecanismo exacto por el que actúan aún se desconoce. Los más conocidos son ivacaftor y lumacaftor19. A pesar de que la llegada de éstos ha mejorado el control de la FQ, se incluyen varias limitaciones que se citan a continuación:
- Necesidad de medicación adicional para el tratamiento de los síntomas.
- Interacción con inductores e inhibidores del CYP3A.
- Efectos secundarios, que incluyen elevación de las transaminasas, cataratas y dolor orofaríngeo.
- Efecto casi nulo en menores de 12 años.
- Necesidad de dosis elevadas, sobre todo en el caso de lumacaftor (se requieren hasta 600 mg).
- Interacción mutua entre lumacaftor e ivacaftor, implicando un aumento del metabolismo del ivacaftor, por lo que se necesitará mayor dosis en el tratamiento combinado.
- Por otra parte, debido a la estructura multi-dominio y al plegado secuencial del CFTR, un único fármaco no puede corregir todos los defectos de los diferentes dominios. Por ello, siempre será necesario utilizar combinaciones. Asimismo, se pueden usar algunos nuevos fármacos para el tratamiento de los síntomas en desarrollo19, como VX-445 y VX-659, que aumentan la expresión del CFTR mediante un mecanismo diferente a los moduladores de primera generación. Estos han demostrado ser eficaces y seguros; sin embargo, el 10% de los pacientes no responden al tratamiento y su coste es muy elevado20.
ADMINISTRACIÓN PULMONAR DE MEDICAMENTOS
La vía pulmonar constituye una ruta muy interesante tanto para el tratamiento local de enfermedades de las vías aéreas como para la administración sistémica de fármacos, sobre todo para aquellos que son poco solubles en medio acuoso o que muestren escasa biodisponibilidad por otras vías, como la oral21. Son muchas las ventajas que ofrece el pulmón como lugar de aplicación. Por ejemplo, su elevada área superficial (aproximadamente 100 m2), el fino epitelio alveolar, la facilidad que tienen los fármacos para atravesar la membrana pulmonar y su extenso flujo (sobre 5 l/min). Todo ello permite la absorción elevada y rápida de muchos fármacos22. Además, la tasa de degradación de los fármacos es baja debido a la escasa actividad enzimática intra y extracelular23. Esto permite que aquellos compuestos con baja magnitud de absorción puedan ser absorbidos con relativa eficacia tras la administración pulmonar.
Por otra parte, para el tratamiento de las enfermedades respiratorias, tras la aplicación pulmonar se alcanza el epitelio pulmonar directamente y, por lo tanto, el sitio de acción, lo que significa que el fármaco actuará inmediatamente. Ello conlleva que las dosis empleadas se reducen significativamente en comparación con otras vías tradicionales como por ejemplo, la oral24.
Sin embargo, a pesar de estas ventajas tan atractivas, los sistemas de inhalación para administrar fármacos no se han empleado muy extensamente25. La posible toxicidad de los fármacos y su degradación por parte de los macrófagos pulmonares, el riesgo de daño pulmonar y el tiempo que los profesionales y los pacientes tendrían que estar expuestos a los fármacos inhalados limitan el uso de esta ruta como vía de administración de fármacos.
Para mejorar la eficacia de los tratamientos de ciertas enfermedades pulmonares y el límite de exposición de ciertos órganos a fármacos potencialmente tóxicos, parece aconsejable facilitar la liberación de los fármacos directamente a los pulmones mediante inhalación. La vía pulmonar proporcionará una liberación específica en la células diana, limitando el tiempo de exposición de las células pulmonares sanas y reduciendo la entrada del fármaco a la circulación sistémica26. Existen en el mercado varios dispositivos de liberación de fármacos que se encuentran suficientemente probados e implementados en la práctica clínica. Sin embargo, otros continúan en fase de desarrollo e investigación.
Surfactante pulmonar
El sistema respiratorio es responsable de la ventilación, asegurada ésta por el proceso cíclico de inspiración-exhalación. También es el encargado de efectuar el intercambio gaseoso de oxígeno y dióxido de carbono entre el medio externo y el alvéolo. El intercambio respiratorio tiene lugar a través de una barrera física relativamente compleja, formada por la capa fina acuosa que rodea al alveolo, las células alveolares epiteliales, la barrera intersticial, las células endoteliales que forman los capilares sanguíneos, el plasma sanguíneo y finalmente la membrana eritrocitaria. Debido al revestimiento líquido del alvéolo, que es consecuencia del metabolismo celular, los pulmones deben cooperar con la tensión superficial de la interfase fluido/aire. Para combatir esta tensión superficial, los neumocitos tipo II secretan surfactante pulmonar, un material especial que se encarga de minimizar las fuerzas mecánicas que llevan a los pulmones a colapsar, sobre todo después del fin de la espiración27. En la Figura 2 se puede apreciar dicha estructura.
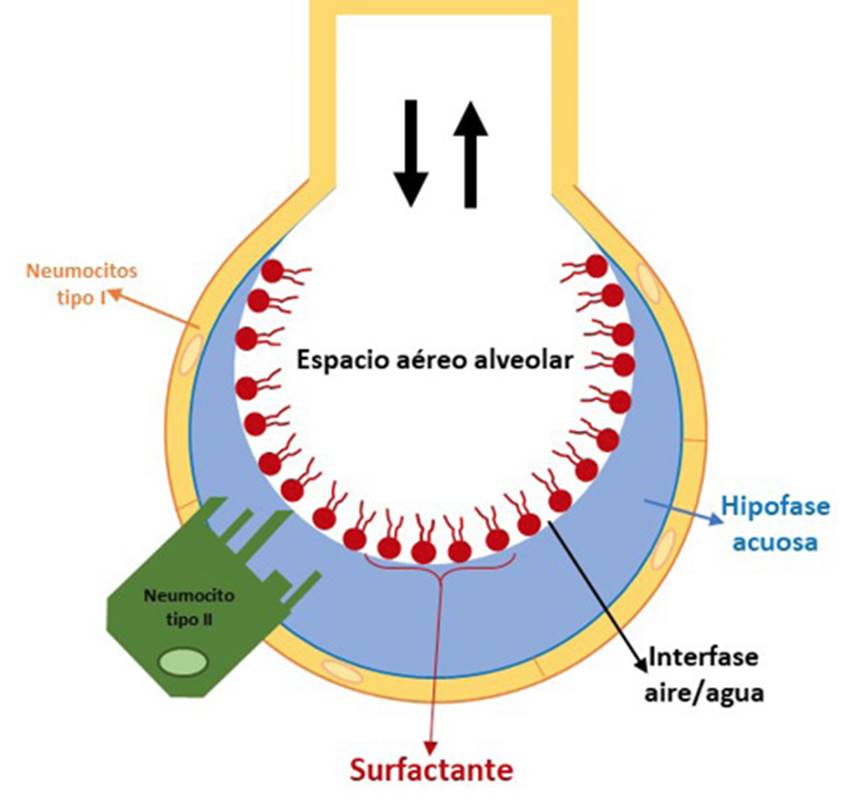
El surfactante pulmonar se compone mayoritariamente de moléculas anfipáticas que forman películas estables en la interfase aire/agua, siendo capaz de reducir notablemente la tensión superficial, desde aproximadamente 70 mN/m en agua pura a temperatura fisiológica hasta un valor mínimo. La razón es que los grupos polares de las moléculas de surfactante establecen interacciones polares con las moléculas de agua interfaciales, reduciendo las fuerzas intermoleculares reticulares cohesivas28. Por lo tanto, siguiendo con el ciclo respiratorio, el esfuerzo de respirar se minimiza durante la inspiración, lo que facilita la exposición de un área bastante elevada para el intercambio gaseoso, mientras la superficie alveolar queda protegida frente al colapso que existe durante el proceso de la espiración.
A la estabilidad de la membrana alveolar contribuyen equitativamente tanto las características del tejido pulmonar como las propiedades del surfactante, lo que se encuentra relacionado con la prevención de diferentes disfunciones respiratorias y patologías29. La deficiencia en el surfactante puede deberse a problemas en el desarrollo pulmonar de ciertos bebés, lo que lleva a causar el síndrome de distrés respiratorio neonatal; asimismo, la inactivación de compuestos activos del surfactante como consecuencia de un daño pulmonar agudo puede contribuir al síndrome de distrés respiratorio, tanto en adultos como en niños. De forma similar, la inactivación del surfactante puede producir cuando el bebé hace su primera inhalación, el denominado síndrome aspiratorio del meconio30.
Se sabe que el surfactante no solo estabiliza la membrana alveolar creando una única bicapa de moléculas anfipáticas en la interfase líquido/aire, sino también estabilizando completamente una red de membranas interconectadas entre la película interfacial y las estructuras de superficie asociadas. Las moléculas que componen el surfactante pulmonar tienen naturaleza anfipática (con grupos hidrófilos y lipófilos) lo que confiere al surfactante sus propiedades activas de superficie31.
Esta composición del surfactante pulmonar define la estructura de su membrana, sus propiedades y funciones. Aproximadamente el 90% del total del surfactante se encuentra constituido por varios tipos de lípidos. Predominan los fosfolípidos, especialmente las fosfatidilcolinas zwitteriónicas (entre el 60 y el 79%), seguidas de especies aniónicas como el fosfatidilglicerol y el fosfatidilinositol, que constituyen el 8-15% del total. Otra fracción importante la componen lípidos neutros, mayoritariamente colesterol en un 8-10%, que es crucial para mantener las propiedades del surfactante y cuyo contenido es clave para optimizar la actividad del surfactante32. En los mamíferos, la especie lipídica más común es la dipalmitoilfosfatidilcolina (DPPC), que se trata de un fosfolípido disaturado que es indispensable para permitir una reducción extrema de la tensión superficial en la interfase alveolar agua/aire. El resto de fosfatidilcolinas y fosfolípidos acídicos son mayoritariamente insaturados, conteniendo ácidos grasos monoenoicos y dienoicos que están esterificados en la posición n-2 del grupo glicerol. Por otro lado, otra fracción importante del surfactante la componen las proteínas, en torno a un 10% del total, en las que se incluyen dos familias: la SP-A (la más abundante) y SP-D, ambas implicadas en mecanismos de defensa innata del alvéolo (hidrofílicas). Asimismo, SP-B y SP-C (hidrofóbicas) son cruciales para la función biofísica del surfactante33.
Deposición de partículas y mecanismos de aclaramiento de las vías aéreas
Una vez administrada la formulación por vía bucal, existen tres mecanismos de deposición para las partículas que van hacia el pulmón: impacto por inercia, sedimentación gravitacional y difusión browniana (Figura 3).

Figura 3. Mecanismos de deposición en las vías aéreas según tamaño de partícula y parte de las vías aéreas alcanzadas. DMMA: diámetro aerodinámico de masa media.
El impacto y la sedimentación adquieren un papel primordial en la deposición de las micropartículas y aglomerados de nanopartículas. Por ejemplo, aquellas micropartículas con diámetro superior a 5 µm se depositan en la orofaringe debido a su gran tamaño. Las partículas de menor tamaño, nanométrico, tienen comportamiento muy diferente ya que no sufren ni impactación ni sedimentación, sino difusión a través de movimientos brownianos que las desplaza hacia zonas distales del árbol respiratorio.
Por otra parte, el aclaramiento mucociliar juega un papel fundamental en el aclaramiento pulmonar de las partículas depositadas por impactación. Las células ciliadas del epitelio mueven el moco y las partículas hacia la faringe, siendo deglutidas o expectoradas21. Es por ello que el aclaramiento mucociliar tiende a tener una mayor importancia en partículas insolubles con un diámetro superior a 6 µm. En cambio, las partículas más pequeñas tienden hacia la deposición alveolar y serán retenidas, o incluso disueltas, en estas áreas pulmonares.
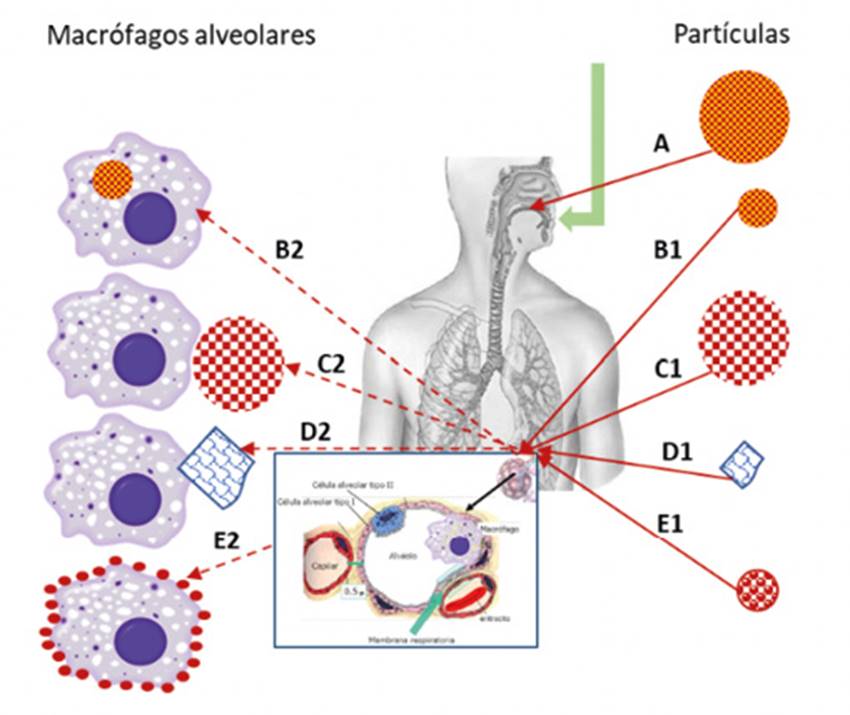
Estas partículas más pequeñas depositadas en el alvéolo pueden seguir diferentes comportamientos según su estructura y composición. Como se recoge en la Figura 4, tras la inhalación, las partículas no porosas con tamaños grandes (normalmente > 5 µm) tienden a quedar retenidas en la orofaringe (A). Partículas no porosas con tamaño inferior (normalmente 1-3 µm) pueden depositarse en alveolos y ser captadas fácilmente por los macrófagos alveolares (B1, B2). Por otra parte, partículas grandes porosas pueden alcanzar niveles distales del pulmón debido a su baja densidad y escapar de la fagocitosis debido a su elevado diámetro (C1, C2). Existen otras partículas en las que su tamaño aumenta significativamente una vez que llegan al pulmón, por lo que el aclaramiento mucociliar se minimiza (D1, D2). Otro tipo de estructuras, como las nanopartículas porosas agregadas pueden, asimismo, alcanzar la zona alveolar, donde se disociaránen nanopartículas que tenderían a ser fagocitadas por los macrófagos alveolares (E1, E2)34.
Otra barrera que dificulta la acción de los fármacos a nivel pulmonar es el aclaramiento por degradación pulmonar. Aunque la actividad metabólica es mucho menor en el pulmón que en el hígado, existen familias del citocromo P450, que actúan a este nivel, siendo las más frecuentes CYP1B1, CYP2B6, CYP2E1, CYP2J2 y CYP3A5 34)1.
Tipos de dispositivos de inhalación
Aunque la terapia inhalatoria resulta de elección para las enfermedades del tipo de la FQ, es un factor crítico el seleccionar adecuadamente el mejor sistema para cada situación concreta, ya que existen algunos inconvenientes que limitan la utilización de uno u otro35. A continuación, se hará una breve descripción de los diferentes dispositivos.
Inhaladores
Los inhaladores constituyen los sistemas más empleados para todo tipo de enfermedades pulmonares, como asma crónica y enfermedades pulmonares obstructivas.
En la 5ª edición de la Real Farmacopea Española, se contemplan tres tipos: inhalador-dosificador presurizado, inhalador-dosificador no presurizado o inhalador de polvo, siendo los sistemas presurizados los más utilizados. Ofrecen múltiples ventajas entre las que se destacan su facilidad de transporte y la posibilidad de liberar una cantidad fija de fármaco. Son frecuentemente empleados para administrar broncodilatadores, corticoides, antiinflamatorios o anticolinérgicos.
Básicamente, los sistemas presurizados están compuestos por gases propulsores, disolventes, solubilizantes, agentes emulsionantes y lubricantes destinados a evitar la obstrucción de la válvula.
La liberación del fármaco tiene lugar al presionar el pulsador una válvula apropiada. Se obtendrá un aerosol (dispersión de partículas sólidas o líquidas en un gas), adaptándose el tamaño de las gotículas o partículas al uso terapéutico previsto. Cambios en el diseño del pulsador y más concretamente en su orificio de salida afectarán sobremanera las características del producto liberado36. La presión necesaria para la liberación de la preparación es generada por los gases propulsores presentes en la formulación.
En la Figura 5 se muestra un esquema de un sistema presurizado.

Figura 5. Partes de un inhalador presurizado. A: pulsador. B: boquilla. C: válvula. D: formulación. Adaptado de Chandel y cols.37.
Los inhaladores de polvo aparecen como alternativa a los sistemas presurizados con el fin de corregir las dificultades de coordinación entre la activación del sistema y el proceso de inhalación. Liberan el contenido en forma de polvo tras ser activados por la inspiración del paciente, lo cual requiere la adecuada colaboración por parte de este. El mecanismo se basa en generar dispersiones del polvo a través las potentes fuerzas que surgen tras la activación del dispositivo. Esto da lugar a una desaglomeración de las partículas hasta individualizarlas. El tamaño de partícula del fármaco estará comprendido entre 1-2 μm, mientras que el de los excipientes empleados para su dilución serán de mayor tamaño, 25-50 μm, para impedir su acceso a las vías aéreas inferiores, ya que impactarán en la orofaringe. Como ejemplo de estos dispositivos se pueden citar el Clickhaler®, el Multihaler®, Diskus®, Spinhaler®y Turbuhaler®, entre otros38. En la Figura 6 se recoge un diagrama representativo de un inhalador típico de polvo.

Figura 6. Inhalador de polvo de cápsulas. A. Pieza de la boca, que sirve para la salida del aire y de la formulación. B. Botón diseñado para activar el dispositivo. C. Filtro / rejilla, que produce cambios en la resistencia interna. D. Cámara de la cápsula. E. Cápsula: la liberación de la dosis después de la compresión de la cápsula tiene lugar a través de la boquilla del dispositivo. F. Salida del aire, que impacta significativamente en la aerosolización del polvo. Adaptado de Chandel y cols.37.
Nebulizadores
Según recoge la Real Farmacopea Española, los nebulizadores son dispositivos que convierten los líquidos en aerosoles mediante gases a alta presión, vibración ultrasónica u otros métodos, obteniéndose tamaños que aseguran el depósito de la preparación en los pulmones
Los nebulizadores pueden ser activados por la inspiración o pueden utilizar otros medios para sincronizar o modificar el funcionamiento del nebulizador con la respiración del paciente. Constituyen un tipo de dispositivos menos empleado39.
Estos sistemas presentan ciertas limitaciones, tales como su utilización con fármacos que posean una solubilidad acuosa limitada, ya que pueden cristalizar o precipitar, haciendo difícil la nebulización. Además, los nebulizadores son menos efectivos que los sistemas presurizados para conseguir una dosis adecuada y una medicación consistente. Por otra parte, la nebulización suele generar más residuos y se desperdicia más fármaco, por lo que los costes se incrementan.
Este dispositivo no requiere coordinación ni pausa respiratoria, y se puede hacer de manera continua, permitiendo administrar diferentes fármacos juntos o por separado, así como modificar la concentración de éstos. La eficacia del dispositivo es variable, según el tipo de nebulizador y de la propia técnica de nebulización.
Básicamente, los nebulizadores pueden ser de dos tipos:
-Tipo “jet” o neumáticos: se usan normalmente para el tratamiento de pacientes con enfermedades pulmonares. Son voluminosos y requieren una fuente generadora de energía (aire comprimido, bombona de oxígeno) (Figura 7). Están constituidos por un reservorio donde se recoge la formulación a nebulizar, un orificio destinado a la entrada del gas y, finalmente, un capilar por donde ascenderá el líquido. La fuerza del gas a presión es la responsable de atomizar el líquido en gotículas de reducido tamaño (1-5 μm)37.

Figura 7. Nebulizador tipo “jet”. A. Aire inhalado adicional. B. Boquilla destinada a la inhalación por parte del paciente. C. Deflector del aerosol que tiene lugar tras el choque del aire con éste. D. Reservorio del fármaco. F. Gas comprimido. Adaptado de Chandel y cols.37.
-Ultrasónicos (Figura 8): son dispositivos que utilizan vibraciones de alta frecuencia para generar el aerosol. Están formados por un cristal piezoeléctrico como sistema generador de ultrasonidos el cual se encuentra conectado a una corriente eléctrica alterna. Dicha energía se transforma en vibración la cual se transmite a la formulación a nebulizar. Como consecuencia de ello se genera una dispersión de pequeñas gotículas (1-3 μm). Debe evitarse el uso de estos sistemas en la formulación de proteínas, suspensiones y antibióticos, debido a que el calor generado puede llegar a desestabilizar o desnaturalizar el preparado35.
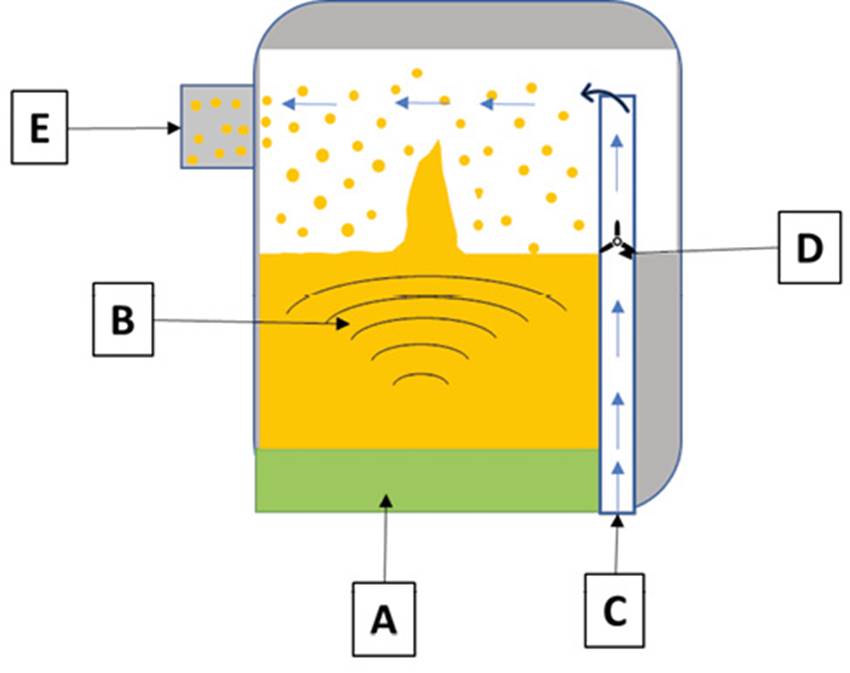
Figura 8. Nebulizador ultrasónico. A. Transductor piezoeléctrico. B. Las ondas ultrasónicas se forman a través del generador. C. Flujo de aire, que se genera a través de la boquilla. D. Ventilador. E. Boquilla de inhalación. Adaptado de Chandel y cols.37.
SISTEMAS MICRO Y NANOPARTICULARES DE ADMINISTRACIÓN PULMONAR
Dado el interés que adquiere el control del tamaño de partícula / gotícula en la efectividad de los sistemas de administración pulmonar, en este apartado se recoge información acerca de los trabajos de investigación desarrollados, aplicando la micro y la nanoencapsulación de moléculas activas.
A modo de resumen, en la Tabla 3 se recopilan algunos de estos sistemas de liberación de fármacos utilizados para administración pulmonar.
Tabla 3. Tipos de micro y nanopartículas empleados para liberación pulmonar de fármacos.
| Sistema | Ventajas | Desventajas | Referencia |
|---|---|---|---|
| Micropartículas convencionales | El proceso de elaboración es sencillo, con técnicas estandarizadas | Fluidificación y dispersión inestables Candidatas a ser fagocitadas por los macrófagos | (40) |
| Grandes micropartículas porosas | Menor tendencia a agregarse y a ser fagocitadas por los macrófagos Alta eficacia de aerosolización | Ausencia de método estandarizado de fabricación con poco control de la eficacia de encapsulación y liberación Dificultad para el escalado | (41) |
| Micropartículas hinchables | Menor fagocitosis por parte de los macrófagos | Fluidificación y dispersión inestables Dificultad para establecer el perfil de liberación del fármaco | (42) |
| Nanopartículas poliméricas | Elevada especificidad Evitan la fagocitosis por los macrófagos y facilitan el transporte transepitelial | Toxicidad pulmonar | (43) |
| Liposomas | Versatilidad en tamaño de vesícula y características físicas Capacidad de incorporar fármacos poco solubles, facilitando la liberación a los macrófagos alveolares, evitando la irritación local | Elevados costes de producción Salida de fármaco debido a la inestabilidad durante la liberación | (44) |
| Nanopartículas sólidas lipídicas | Biocompatibles y más estables que los liposomas para la nebulización, facilidad de escalado e inocuas | Menor capacidad de encapsular fármacos que los liposomas Perfil de liberación inestable Posibilidad de solidificar | (45) |
| Partículas porosas- agregados de partículas | Disminución de la agregación y elevada eficacia de aerosolización Elevada especificidad Escapan del aclaramiento de los macrófagos alveolares y facilitan el transporte transepitelial | No son fácilmente redispersables para su administración | (46) |
En general, las nanopartículas representan la globalidad de las partículas con tamaño inferior a 500 nm, y se administran usualmente como dispersiones coloidales estables. Se utilizan como sistemas de administración de fármacos por diversas vías, entre ellas la inhalatoria. Durante el proceso de formación del sistema disperso en el que van incluidas, las nanopartículas pueden formar agregados de mayor tamaño, o ir incluidas en gotículas, ambos de tamaño micrométrico. Su composición, forma y tamaño influirán posteriormente en la deposición y en el tiempo de residencia en el pulmón40.
Dentro de la amplia variedad de formulaciones nanoparticulares existentes, nos centraremos en los sistemas de naturaleza lipídica de administración por vía pulmonar entre los que cabe destacar los liposomas, las nanopartículas sólidas lipídicas y las nanopartículas híbridas de lípidos-polímeros.
Los liposomas son, probablemente, los sistemas más usados y mejor caracterizados. Son vesículas constituidas por una bicapa lipídica (unilaminares) o varias (multilaminares), que encierran un espacio acuoso.
Han demostrado tener un futuro prometedor en el tratamiento de las patologías pulmonares debido a las siguientes razones:
- Capacidad de solubilizar fármacos poco solubles.
- Pueden proporcionar liberación sostenida a lo largo del tiempo, lo cual prolonga los niveles terapéuticos locales y sistémicos.
- Evitan la irritación pulmonar.
- Facilitan la liberación intracelular de fármacos, principalmente porque son captados por los macrófagos.
- Capacidad de llegar a dianas específicas usando ligandos de superficie o anticuerpos.
- Potencial para ser absorbidos a través del epitelio intacto para alcanzar la circulación sistémica.
En la Figura 9 se muestra la localización de los liposomas en diferentes órganos una vez son administrados por vía intravenosa o vía inhalatoria. Se puede apreciar la distribución específica hacia los pulmones tras una administración por esta vía, a diferencia de un mayor reparto a diferentes órganos tras la administración intravenosa.

Figura 9. Distribución de los liposomas en los diferentes órganos según vía de administración. Adaptado de Kuzmov y Minko47.
Otra razón que justifica su elección para ser utilizados por esta vía es la similitud que existe entre la composición química del surfactante y la de los liposomas. Como se ha mencionado anteriormente, el surfactante pulmonar está formado por una mezcla 90/10 de lípidos/proteínas. A su vez, el 70% aproximadamente de la parte lipídica está constituido por DPPC o fosfatidilcolina.
Entre los mecanismos disponibles para la liberación pulmonar, la nebulización es la técnica más empleada para administrar liposomas48. Para soslayar los problemas de inestabilidad a largo plazo que presentan las formulaciones convencionales es muy frecuente transformar los preparados liposomales en polvos secos, con o sin excipientes, a través de diferentes técnicas como la liofilización, secado por pulverización mediante frío o mediante tecnologías de fluidos supercríticos.
Las nanopartículas sólidas lipídicas son estructuras esféricas, con diámetro comprendido entre 50 y 500 nm, que están constituidas por un lípido sólido rodeado de una monocapa de tensioactivo, usualmente fosfolípidos.
Una de las recientes aplicaciones de este tipo de estructuras ha sido desarrollada por Rosière y cols. quienes, seleccionando lípidos similares a los que contiene el surfactante pulmonar, elaboraron nanopartículas de paclitaxel para terapia inhalatoria, incluyendo DPPC/DPPE-PEG en su superficie49. Estas estructuras lipídicas mostraron elevada capacidad de carga con gran potencial para la quimioterapia inhalatoria.
Las nanopartículas híbridas de lípidos-polímeros son nanopartículas poliméricas recubiertas de un lípido, habitualmente un fosfolípido50. Un ejemplo de este grupo lo constituyen las nanopartículas de ácido poliláctico-glicólico recubiertas de fosfatidilcolina o fosfatidilcolina-estearilamina, las cuales han sido probadas como híbridos de lípido-polímero para la terapia inhalatoria.
Otro híbrido polímero-lipídico de nanopartículas, adecuado para la terapia inhalatoria recibe la denominación de “Janus”, que ha sido desarrollado por Garbuzenko y cols.51. La principal aportación de esta formulación se basa en la posibilidad de añadir dos compuestos de naturaleza muy diversa en la misma nanopartícula. Concretamente, dichos autores consiguieron incorporar simultáneamente clorhidrato de doxorubicina en la fase polimérica acuosa y curcumina, fármaco insoluble, en la fase lipídica, manteniendo adecuadas propiedades de tamaño, forma y carga de las partículas.
AVANCES EN EL USO DE LIPOSOMAS PARA EL TRATAMIENTO DE LA FIBROSIS QUÍSTICA
En este último apartado se hará una breve exposición de las formulaciones en liposomas que se pueden utilizar en el tratamiento de la FQ. Previamente, en la Tabla 4, se recoge un resumen de diferentes proyectos, entre los cuales se incluyen además algunos basados en nanopartículas, por su especial interés, centrados la mayoría de ellos en mejorar la sintomatología de esta enfermedad. Algunas de las formulaciones descritas se encuentran ya comercializadas y otras en fase de investigación.
Tabla 4. Recopilación de estudios en los cuales se usan nanotransportadores para tratar la FQ.
| Fármaco | Enfermedad que trata | Tipo de sistema de nebulización | Tipo de nanosistema | Resultados obtenidos | Referencia |
|---|---|---|---|---|---|
| Amikacina Arikace®, INSMED | Tratamiento de infecciones crónicas de Pseudomonas aeruginosa | Nebulizador e inhalador | Liposomas | Mejora de infecciones crónicas de pulmón | (52) |
| Amikacina eFlow® | Tratamiento de infecciones crónicas de Pseudomonas aeruginosa(liberación sostenida) | Nebulizador | Liposomas | Mejora de infecciones crónicas de pulmón | (53) |
| Amikacina | Infección pulmonar por Pseudomonas aeruginosa | Suspensión para inhalación | Liposomas | Disminución de la densidad del esputo y mejora de síntomas respiratorios | (54) |
| Calcifediol | Prevención de la enfermedad pulmonar provocada por Pseudomonas aeruginosa | Nebulizador tipo jet | Liposomas | Los excipientes demostraron no tener efectos adversos y el fármaco se presentó como una alternativa para prevenir la infección | (55) |
| Ciprofloxacino (Pulmaquin®y Lipoquin®) | Terapia de mantenimiento y mejora del tratamiento antimicrobiano para Pseudomonas aeruginosa | Nebulizador | Liposomas | Los datos muestran una mejora en el control de la liberación del ciprofloxacino | (56) |
| Ciprofloxacino | Conseguir la liberación sostenida del fármaco para el tratamiento de infecciones pulmonares | Aerosol | Nanocristales en liposomas | Liberación prolongada de ciprofloxacino desde las formulaciones | (57) |
| Colestimetato de sodio (Colomycin®) | Mejora de la terapia antimicrobiana para Pseudomonas aeruginosa | Nebulizador de malla vibrante | Nanopartículas sólidas lipídicas Sistemas nanoestructurados | Las partículas mostraron una buena distribución in vivomejorando el tratamiento antibacteriano | (58) |
| Gentamicina | Tratamiento de infecciones crónicas de Pseudomonas aeruginosa | Aerosol | Liposomas | El tratamiento con las formulaciones de liposomas podría ser más efectivo que el fármaco solo | (59) |
| Galio-Gentamicina | Tratamiento de infecciones crónicas de Pseudomonas aeruginosa | In vitro | Liposomas | La presencia de galio reduce la formación de la biopelícula, incrementando así la eficacia del tratamiento | (60) |
| Lumacaftor e ivacaftor | Mejora y regulación del gen/proteína CFTR aumentando el número de canales y su amplitud | Inhalador | Sistemas lipídicos nanoestructurados | Mejora de la regulación, con respecto a los dos fármacos sin encapsular | (19) |
| Plásmido de ADN que codifica el gen CFTR | Restauración de la función de la proteína CFTR | Nebulizador activado por respiración | Liposomas | Estabilización de la función pulmonar | (61) |
| Tobramicina | Infecciones del tracto respiratorio provocadas por Pseudomonas aeruginosa | Nebulizador | Sistemas lipídicos nanoestructurados | La alternativa de la encapsulación ofrece mejoras significativas en el tratamiento en comparación con el fármaco en solitario | (62) |
| Vector plásmido (pGM169) | Ha reducido la inflamación en cobayas | Aerosol | Complejo con lípido catiónico | Las dosis empleadas mostraron pocos efectos adversos, reduciendo la inflamación; futuro uso en humanos. | (63) |
La observación de muestras tisulares de pacientes con FQ sugiere que Pseudomonas aeruginosa se localiza predominantemente a nivel intraluminal, distribuyéndose en masas mucopurulentas hipóxicas con una composición compleja. Tanto dicha composición como el modo de crecimiento de la biopelícula por parte de la bacteria constituyen un desafío difícil para la terapia con antibióticos.
En el caso de los aminoglucósidos, hay que destacar su lenta capacidad de penetración debido a las interacciones electrostáticas con las matrices de moco y con la biopelícula. Esto da lugar a concentraciones subinhibitorias de estos fármacos que no impiden la formación de la biopelícula. La posibilidad de aumentar la eficacia administrando directamente estos antibióticos a los pulmones por inhalación se convierte en una alternativa. Sin embargo, debido al tamaño molecular relativamente pequeño de estos fármacos, se eliminan rápidamente de los pulmones después de la inhalación. Esto hace que la mayor parte del tiempo se encuentren a nivel local en niveles inferiores a la concentración mínima inhibitoria efectiva. Además, debido a su corto tiempo de residencia en pulmón, estos medicamentos requieren una administración de, al menos, dos veces al día.
En base a estas consideraciones previas, se están efectuando numerosos estudios de cara a lograr una mejora en la terapia con aminoglucósidos mediante la administración de dosis suficientes para mantener niveles sostenidos del mismo en la zona a tratar.
En este sentido, el uso de liposomas inhalados para administrar el antibiótico de manera localizada y sostenida se ha extendido en los últimos años con importantes aportaciones. Estas formulaciones liposomales se diseñan para proporcionar una liberación controlada o sostenida del fármaco encapsulado y reducir la absorción sistémica, prolongando así el tiempo de residencia del fármaco en el pulmón. Dicho perfil de liberación mantendrá elevadas concentraciones del antibiótico a nivel local (por encima de la concentración mínima inhibitoria), reduciendo así la frecuencia de administración. Además, los macrófagos pueden fagocitar los liposomas cargados con el fármaco, lo que permitiría el tratamiento de infecciones intracelulares, como las causadas por micobacterias no tuberculosas.
Dado que los aminoglucósidos son antibióticos policatiónicos, se pueden proteger mediante su encapsulación en liposomas, vesículas lipídicas biodegradables, de su inactivación por los componentes polianiónicos presentes en el esputo, tales como mucinas o ADN. Con este fin, se ha desarrollado una amikacina liposomal, que se administra por inhalación (Arikace®, INSMED), consistente en liposomas con carga neutra (Figura 10). Esta formulación ha sido diseñada para mejorar la penetración del aminoglucósido en el moco y en las biopelículas de Pseudomonas aeruginosa52.Tras su administración mediante el dispositivo nebulizador eFlow®, se apreció que se producía una liberación sostenida en los pulmones a través de la evaluación en estudios en 24 pacientes con FQ e infección crónica por esta bacteria53. Los voluntarios recibieron 500 mg de Arikace una vez al día durante 14 días. Los ensayos clínicos de fase II randomizados, con control de placebo en pacientes con infección crónica de Pseudomonas aeruginosacon una dosis de 560 mg una vez al día durante 28 días consecutivos, seguidos de 28 días de descanso, mostraron un beneficio clínico mayor que con el placebo, reduciendo la densidad del microorganismo y mejorando su función pulmonar54. Además, el medicamento fue bien tolerado.
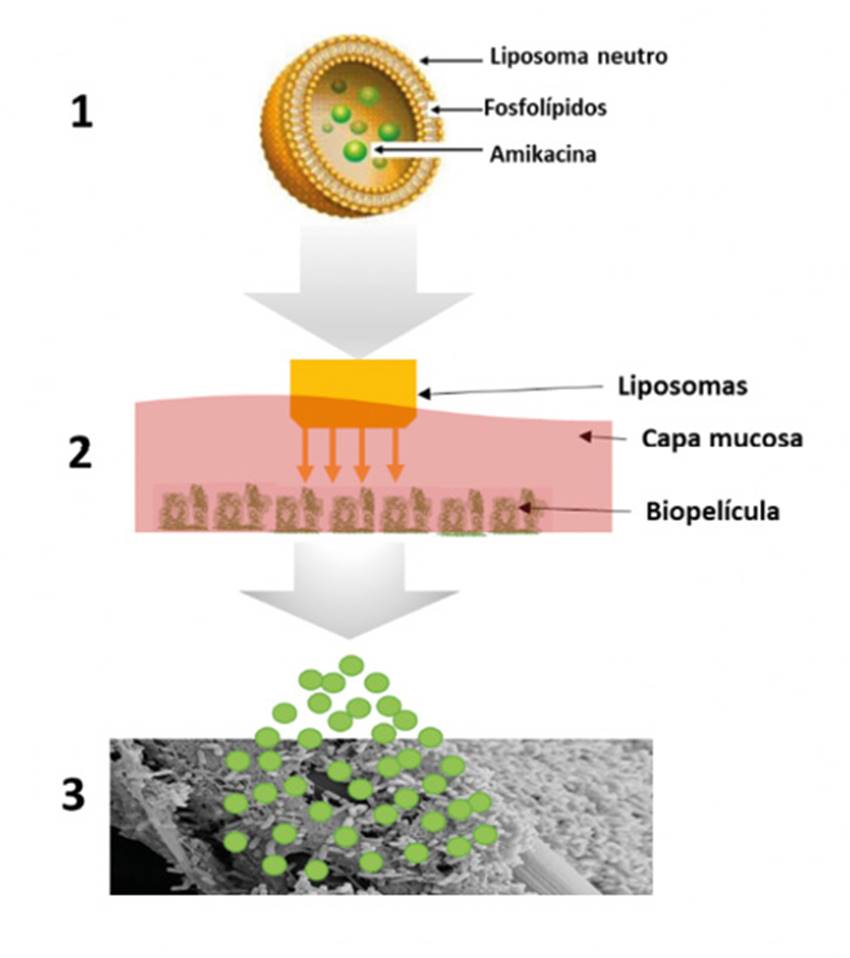
Figura 10. Mecanismo de acción de Arikace®. 1: el fármaco policatiónico se encuentra dentro del núcleo hidrofílico del liposoma. 2: la barrera pulmonar y el moco de las vías aéreas presentan carga negativa, mientras que Arikace®tiene carga neutra, lo que permite la rápida circulación a través de la biopelícula y del moco. 3: una vez que el liposoma llega al sitio de acción, las bacterias liberan una serie de agentes que provocan la lisis del liposoma y, por lo tanto, la salida del antibiótico. Adaptada de Clancy y cols.64.
En los últimos años se han planteado diversos estudios que establecen una conexión entre la vitamina D3 y enfermedades pulmonares, tales como el asma y la enfermedad pulmonar obstructiva crónica, como consecuencia de su papel en la regulación del sistema inmune. Asimismo, las deficiencias en los niveles séricos de esta vitamina, específicamente en pacientes con FQ, probablemente como resultado de malabsorción, hace que se plantee la hipótesis de que la administración de vitamina D3 o sus metabolitos directamente al pulmón sea muy beneficiosa. Sin embargo, la escasa solubilidad en agua de estos compuestos requeriría su disolución en solventes como el etanol, lo que limita su administración por vía inhalatoria. Es por ello por lo que se han desarrollado liposomas de calcifediol, mostrándose en los estudios una mejora significativa en la acción antibacteriana55.
Ciprofloxacino es una fluoroquinolona que tiene un potente efecto bactericida sobre la biopelícula de Pseudomonas aeruginosa, ya que afecta a la función de la enzima girasa en esta bacteria. Este fármaco ha sido ampliamente utilizado por vía oral e i.v. en pacientes con FQ y con otras patologías si bien, por estas vías, se consiguen bajas concentraciones a nivel pulmonar. El uso de dosis más elevadas con el fin de aumentar los niveles de fármaco localmente conduciría a la aparición de importantes efectos secundarios sistémicos. De ahí surge el interés por desarrollar formulaciones inhaladas de este fármaco65.
Aprovechando las ventajas que ofrecen los liposomas para administrar fármacos por vía inhalatoria, se han realizado estudios con esta fluorquinolona.
Aparecen en la literatura dos tipos de formulaciones de ciprofloxacino de gran interés, que se diferencian fundamentalmente en el método de preparación de los liposomas:
-Elaborados mediante extrusión de liposomas multilaminares a través de filtros de membrana de 80 nm, seguido de encapsulación de ciprofloxacino por carga remota56
-Elaborados mediante el proceso de congelación y calentamiento (Frozen and Thawed), seguido de atomización por spray drying57.
El primer método se utiliza en la fabricación de Lipoquin®, que tiene un 99% del ciprofloxacino encapsulado, y de Pulmaquin®(ambos de Aradigm Inc., Hayward, CA), con un 70% de ciprofloxacino encapsulado y el 30% restante sin encapsular. Ambas formulaciones han demostrado ser muy prometedoras para el tratamiento vía inhalatoria de la FQ. Como el ciprofloxacino no encapsulado es poco soluble a pH 6, por ello se preparan dos viales de esta formulación, uno con este fármaco encapsulado en liposomas y otro con ciprofloxacino libre. Esto da lugar a una liberación inmediata por parte del fármaco libre, seguida de una liberación sostenida durante 24 horas del ciprofloxacino liposomal. Estas formulaciones han demostrado ser eficaces para tratar las infecciones pulmonares por Yersinia pestisy la fiebre Q. Ambos preparados pudieron ser aerosolizadas perfectamente, manteniendo la integridad del liposoma, aunque en algunos lotes se observó, a largo plazo, la formación de cristales del fármaco, lo que podría reducir su estabilidad56.
Mediante el segundo método se obtienen formulaciones de liposomas para ser administradas en forma de polvos inhalables de nanocristales del fármaco, encapsulados en liposomas, con el fin obtener perfiles de liberación prolongada. El proceso de atomización permite, además, aumentar la estabilidad del preparado57.
En lo que respecta a gentamicina, algunos estudios indican que cuando se incorpora en liposomas mejora su efectividad frente a determinados microorganismos como es el caso de Pseudomonas aeruginosa, disminuyendo su resistencia, tal como recogen Mugabe y cols. en sus trabajos59.
Con el fin de incrementar la eficacia antibacteriana de gentamicina, se formuló conjuntamente con galio, un agente inhibidor eficaz contra el crecimiento de Pseudomonas aeruginosay la formación de biopelículas. Debido a su similitud química con el hierro, puede sustituirlo e inhibir procesos biológicos dependientes del mismo, que resultan en un incremento de la vulnerabilidad de la mayoría de las bacterias infecciosas. Estudios in vitrohan demostrado que la formulación conjunta de galio-gentamicina fue capaz de eliminar las biopelículas de esta bacteria y bloquear la comunicación bacteriana (quorum sensing), empleando concentraciones muy bajas de gentamicina (0.94 mg/l), siendo además más eficaz que la que contenía el antibiótico en solitario. El galio sigue siendo investigado por su potencial para el tratamiento de infecciones bacterianas en la FQ60.
Otro enfoque diferente en el tratamiento de esta enfermedad se basa en tratar la causa fundamental de la misma, dirigiéndose hacia la disfunción de la proteína CFTR2)(63. Una de las estrategias consiste en la transferencia génica a nivel pulmonar, siendo la clave del éxito la interacción adecuada entre el agente de transferencia génica y el dispositivo de nebulización. A este respecto existen estudios muy prometedores que evalúan los efectos de la aerosolización en una formulación de un plásmido de ADN (pDNA) complejado con el liposoma catiónico GL67A (pDNA/GL67A) utilizando dispositivos nebulizadores disponibles comercialmente. Los resultados mostraron la resistencia del plásmido durante el proceso de administración, siendo estas formulaciones objeto de futuros estudios clínicos de fase IIa / b en pacientes con FQ63.
CONCLUSIONES
- Actualmente, los tratamientos para la FQ se basan en intentar mejorar la sintomatología. Por ello, se hace interesante desarrollar en el futuro fármacos que traten el defecto en el gen CFTR, origen de la enfermedad.
- La vía pulmonar es idónea para administrar medicamentos que traten patologías que cursen a este nivel.
- La Nanotecnología se ofrece como alternativa al tratamiento de la FQ por su capacidad de administrar nanotransportadores por vía pulmonar y liberar de forma localizada y sostenida su contenido.
- Los liposomas pueden fabricarse a base de lípidos biocompatibles, similares a los que componen la membrana y el surfactante pulmonar. Estas formulaciones pueden ser aerosolizadas fácilmente y son bien captadas por los pulmones, permitiendo una retención más prolongada.
- Uno de los principales problemas a resolver en el tratamiento de la FQ es la eliminación de la biopelícula ya que esta estructura dificulta la acción del antimicrobiano. Uno de los principales desafíos que se deben ejecutar en este campo será el desarrollo de formulaciones liposomales adecuadas que a través de su composición favorezcan la destrucción de esta biopelícula.

