










 Servicios personalizados
Servicios personalizados
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkHighlights
Topically applied drugs can be delivered as proniosomal vesicular system.
Vesicular system can be used to increase the penetrability of the drug.
Span, soy lecithin and cholesterol can be used to develop stable formulation.
Statistical technique like central composite design would be helpful in formulation optimization.
Introduction
Non-steroidal anti-inflammatory drugs (NSAIDs) are frequently prescribed for arthritis, low back pain and some joint diseases1. The well reported mechanism of action is reversible inhibition of cyclooxygenase enzyme (COX) and decrease the synthesis of prostaglandins. However, due to inhibition of prostaglandins (PGs) which protects the gastric mucosa, they show side effects including dyspepsia to peptic ulcer and gastrointestinal haemorrhage. NSAIDs are acidic in nature they may produce local irritation and lesions on the gastrointestinal mucosa. Hence, some of the NSAIDs are administered percutaneously and transdermally to achieve local or systemic effect as an alternative to oral and parenteral administration2. However, the barrier layer i.e. stratum corneum prevents the penetration of the drugs to lower layers of the skin and/or to enter systemic circulation. In this context, the formulation plays a key role in the penetration and absorption of the active ingredient.
Etodolac is BCS Class II drug. It is a selective COX-2 inhibitor with 10-fold COX-2 selectivity over COX-1; therefore, it can be prescribed safely for the treatment of acute pain and inflammation. It has poor water solubility (75 µg/ml) because of high hydrophobicity3. It causes gastric irritation, constipation, diarrhoea, vomiting, headache dizziness, sore throat and blurred vision4) and may create limitation in formulation of oral dosage forms. By considering the drawbacks of oral delivery of etodolac; it would beneficial to give it through topical route using novel vesicular drug delivery system. It was reflected from the literature that niosomes; a vesicular system could be beneficial for topical and transdermal delivery because they act as a reservoir of drug for a prolonged period of time and enhance skin penetration5,6 and thus increases bioavailability. However the niosomes are also has instability issues like fusion, aggregation, sedimentation and leakage of entrapped drug. These mentioned problems can be avoided by using non ionic surfactant formulation which is anhydrous, free flowing i.e. proniosomes and can form vesicles after hydration (niosomes). Proniosomes can entrap both hydrophilic and lipophilic drugs. Proniosomal gel is beneficial to reduce drugs associated side effects, increase effectiveness and make the formulation physically stable. In this context we had considered development of vesicular topical formulation with optimal quality in short period of time with less number of experimental trials using varying proportion of soy lecithin, cholesterol and non-ionic surfactant7. Hence, two level three factor rotatable central composite design (CCD) was used study the effect of amount of soy lecithin, non ionic surfactant and cholesterol on entrapment efficiency (%EE) and % drug release.
Materials and Methods
Materials
Etodolac was received as a gift sample from Swiss Garnier Life Sciences, Una, Himachal Pradesh, India. Lipoid S 100 and Phospholipon 80H were obtained from Ludwings Shafen, Germany. Span (20, 40 and 60) was purchased from S. D. Fine Chem. Ltd., Mumbai, India. Cholesterol was obtained from Thomas Baker Ltd., Mumbai, India. All other chemicals used in this study were of analytical grade.
Methods
Preformulation study
To eliminate the possibility of interaction between etodolac and cholesterol, lecithin and Span, compatibility study was carried out by storing the physical mixture (1:1) in the stability chamber at 40 °C/75% RH for 1 month. At the end of the study, FT-IR spectrums of etodolac alone and binary mixture were recorded and studied.
Prior incorporation of etodolac, trial formulation batches containing varying proportions of Span (20, 40 and 60): lecithin (Lipoid S 75, Phospholipon 80 H, Lipoid S 100) in ratio of 0.5:1, 1:1, 1:0.5 and 1:0.75 were developed. These preparations were observed for homogeneity, consistency, clogging and sudden change in viscosity.
Development of proniosomal gel
Etodolac proniosomes were prepared by modified coacervation phase separation method reported by Thakur et. al8. Briefly, etodolac (5% w/w) and varying concentrations of Span 60, Lipoid S 100 and cholesterol were mixed with 2.5 ml of absolute ethanol in a wide mouth glass tube. The ingredients were mixed together; open end of the glass tube was covered and warmed on a water bath at 60-70 ºC for about 5 min. Phosphate buffer saline (pH 7.4) was added and warmed on a water bath for ≈2 minutes to form dispersion. The mixture was allowed to cool to room temperature until the dispersion was converted to proniosomes having semisolid consistency and stored in dark until further use.
Optimization by design of experiment
A full central composite design (rotatable) was used in this study. Design matrix is given in Table 1. The amount of Span 60 (A), Lipoid S 100 (B) and cholesterol (C) were taken as independent variables, while % entrapment efficiency (Y1) and % drug release (Y2) were taken as dependent variables. The factors were studied at 2 levels (−1, +1) indicating low and high respectively. The design consisted of three groups of design points, including two level three factor factorial design points, axial or star points and center points. Total 20 experiments consisting of 8 factorial points, 6 axial points, and 6 replicated central points were performed according to the central composite design matrix generated by Design Expert V. 11 Software (Table 2) (Stat-Ease, Minneapolis, USA).
Table 1: Two level three factor central composite design
| Factor | Low level | High level | -α | + α |
|---|---|---|---|---|
| Experimental domain (Coded values) | ||||
| Span 60 (mg) | -1 | +1 | -1.682 | +1.682 |
| Lipoid S 100 (mg) | -1 | +1 | -1.682 | +1.682 |
| Cholesterol (mg) | -1 | +1 | -1.682 | +1.682 |
| Experimental domain (Actual values) | ||||
| Span 60 (mg) | 1200 | 1800 | 995.46 | 2004.5 |
| Lipoid S 100 (mg) | 500 | 1000 | 329.55 | 1170 |
| Cholesterol (mg) | 50 | 100 | 32.96 | 117.04 |
Table 2: Experimental design batches of proniosomes
| Experiment | Span 60(mg) | Lipoid S 100 (mg) | Cholesterol (mg) | Ethanol(ml) | Saline sol.(ml) |
|---|---|---|---|---|---|
| Factorial designs | |||||
| CD 1 | 1200 | 500 | 50 | 2.5 | 1.8 |
| CD 2 | 1800 | 500 | 50 | 2.5 | 1.8 |
| CD 3 | 1200 | 1000 | 50 | 2.5 | 1.8 |
| CD 4 | 1800 | 1000 | 50 | 2.5 | 1.8 |
| CD 5 | 1200 | 500 | 100 | 2.5 | 1.8 |
| CD 6 | 1800 | 500 | 100 | 2.5 | 1.8 |
| CD 7 | 1200 | 1000 | 100 | 2.5 | 1.8 |
| CD 8 | 1800 | 1000 | 100 | 2.5 | 1.8 |
| Star design | |||||
| CD 9 | 995.46 | 750 | 75 | 2.5 | 1.8 |
| CD 10 | 2004.5 | 750 | 75 | 2.5 | 1.8 |
| CD 11 | 1500 | 329.55 | 75 | 2.5 | 1.8 |
| CD 12 | 1500 | 1170.45 | 75 | 2.5 | 1.8 |
| CD 13 | 1500 | 750 | 32.96 | 2.5 | 1.8 |
| CD 14 | 1500 | 750 | 117.04 | 2.5 | 1.8 |
| Center points | |||||
| CD 15 | 1500 | 750 | 75 | 2.5 | 1.8 |
| CD 16 | 1500 | 750 | 75 | 2.5 | 1.8 |
| CD 17 | 1500 | 750 | 75 | 2.5 | 1.8 |
| CD 18 | 1500 | 750 | 75 | 2.5 | 1.8 |
| CD 19 | 1500 | 750 | 75 | 2.5 | 1.8 |
| CD 20 | 1500 | 750 | 75 | 2.5 | 1.8 |
Evaluation of proniosomal gel
Determination of pH and viscosity
The pH of all formulations was determined by using pH meter at ambient temperature. The pH meter was calibrated before each use. About 1g of gel was dissolved in 25ml of distilled water and pH was measured by immersing the electrode. The viscosities of different proniosomal gel formulation were determined at 25°C using a Brookfield viscometer. The formulation (more than 5g) was placed in a beaker and was allowed to equilibrate for 5 min before measuring the dial reading using a spindle no S-64 at 20 rpm6,9.
% Encapsulation efficiency (%EE)
Approximately 0.2 g of proniosomal gel was dispersed in 10 ml of phosphate buffer (pH 7.4). The dispersion was sonicated, centrifuged at 18,000 rpm at -20°Ċ for 30 min. (C-24 BL, Remi lab) to separate unentrapped drug. The supernatant was removed and quantified by an UV spectrophotometer at 278 nm10. The % encapsulation was calculated by the following equation:
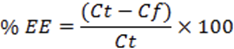
Where,
Ct is the concentration of total etodolac; Cf is the concentration of free etodolac.
Vesicle size and zeta potential
Vesicle size, size distribution and zeta potential were determined by Malvern Zetasizer in order to ascertain the stability as well as the reproducibility of the method. About 1g of gel was dissolved in 20 ml double distilled water and agitated to get homogeneous dispersion. Mean globule diameter and zeta potential was recorded11.
Rate of Spontaneity
This test helps to determine the numbers of niosomes formed spontaneously after hydration in 15-20 min. Approximately 100 or 200 mg of proniosomal gel was transferred to glass bottle and spread uniformly around walls. About 20ml of saline solution was added along the walls and was left aside for 20 min. then a drop was withdrawn and place on Neubauers chamber to count the number of vesicles12.
In-vitro and Ex-vivo drug release
In-vitro release study was performed by using diffusion cell apparatus and egg membrane. Egg membrane was separated by putting the egg shell in mixture of water and hydrochloric acid in which the shell is dissolved to calcium chloride with effervescence of carbon dioxide. Before initiation of study; the egg membrane was soaked in phosphate buffer for 24 hours. About 1g of proniosomal gel was placed in the donor compartment tube and spread over the egg membrane which was clamped at the end of the tube. The acceptor compartment consists of phosphate buffer (pH 7.4). The entire surface of the cell was in contact with the medium present in the receptor compartment which was agitated by magnetic stirrer at rpm 600 and temperature 32±1°Ċ. A sample was removed at specific time interval for up to 24 h with same amount replaced to maintain sink condition. The sample was analysed at 278 nm against blank using UV spectrophotometer. For ex-vivo study; the egg membrane was replaced by excised skin of abdominal region of rat. The stratum corneum of the skin faced the drug donor compartment whereas dermis faced the receiver compartment13-16.
Scanning electron microscopy
The surface morphology of the formulation was determined by placing the drop of niosome dispersion on carbon coated grid, vacuum dried and observed microscopically at an accelerating voltage of 10 Kv using scanning electron microscope (JEOL/JEM 2100, Japan).
Anti-inflammatory study
The study protocol was approved by the Animal Ethical Committee of MET'S Institute of Pharmacy (CPCSEA Reg. no-1344/ac/10/CPCSEA/IAEC) and it was conducted as per the guidelines. Fifteen albino mice were selected. They were divided into 3 groups (control, standard and test) each containing 5 animals and kept fasted overnight. About 0.1ml of 1 % (w/v) carrageenan was injected on the left hind paw to induce edema. Control group received carrageenan (1% w/v) only while standard and test were treated with marketed and developed proniosomal gel in addition to carrageenan. The paw thickness was measured using vernier calliper at the time interval of 0, 30, 60, 120 and 180 minute. The % inhibition of diameter paw edema by proniosomal gel and marketed gel treated group compared with carrageenan control group17. From the mean edema volume, the percentage inhibition of the edema was calculated between the treated and control groups. Before anti-inflamatory activity skin irritation study was conducted to find out the allergic potential of formulation.


Where,
Vc = Paw volume of control
Vt = Paw volume of test
Vs = Paw volume of standard
Results and Discussion
Preformulation study
The FT-IR spectrums of etodolac, etodolac with Span 60, Lipoid S-100 and cholesterol are shown in Figure 1. The principle peaks of the etodolac i.e. -NH stretching at 3342.64 cm-1, -CH stretching in alkane at 2970.38 cm-1 and -CO stretching in ketone at 1745 cm-1, -CO stretching in aromatic at 1261 cm-1 were present in all physical mixtures. There was neither deletion of existing peak nor addition of new peak in the spectrum revealed the compatible nature of the excipients with etodolac. Hence; by using these materials stable formulations were developed.
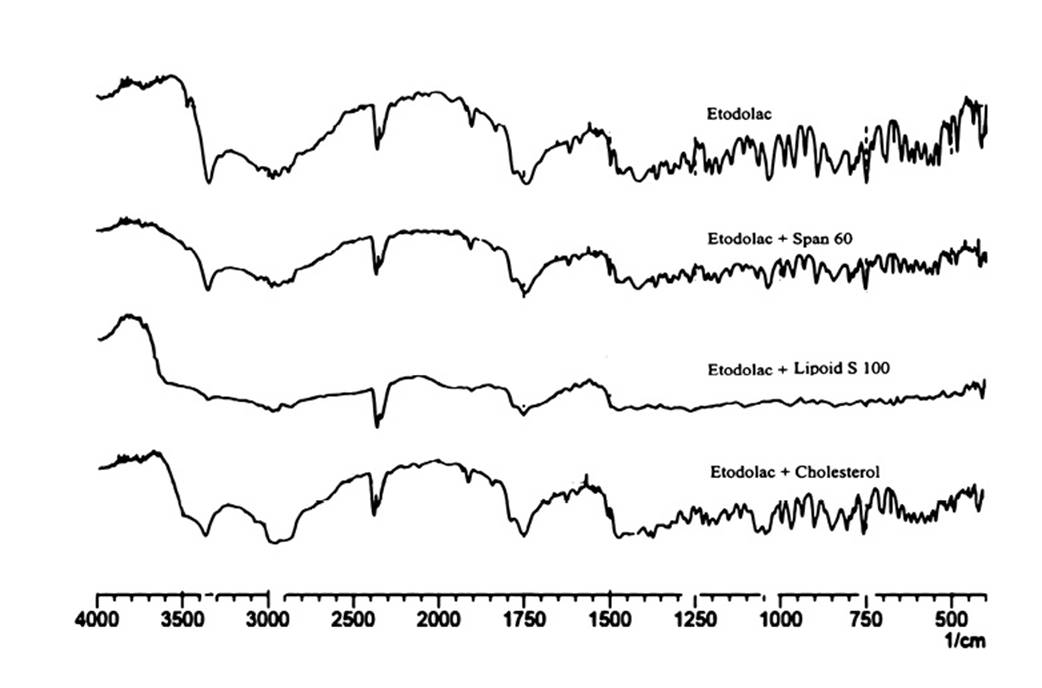
Figure 1. FTIR spectrums for prediction of incompatibility: etodolac and its binary mixture with Span 60, Lipoid S 100 and cholesterol.
Preliminary studies have demonstrated that formulations of Span 20 and 40 with Lipoid S 100 and Phospholipon 80 H were transparent; yellow and semisolid. Gels of Span 40 had shown clogging with change in viscosity after 1 week of storage. In case of Span 20, formulations were stable but less amount of etodolac was incorporated. Hence; it would be beneficial to use Span 60 as it had shown high entrapment efficiency in addition to consistency. Similarly niosome vesicles prepared with Lipoid S 100 were observed good and denser than prepared with Phospholipon 80 H.
Data analysis by central composite design
Effect of composition variable on %EE
Proniosomes are made of lipidic materials and considered as it has ability to entrap drug. The response (Y1) i.e. %EE was ranged from 74.36±0.22 to 90.85±0.26 (Table 3). ANOVA test revealed that the response surface quadratic model was significant. The polynomial equation for encapsulation efficiency is
%EE=+81.42-1.58*A-2.35*B+1.75*C-2.19*A*B-2.30*A*C+0.96*B*C-0.88A2-0.13B2+0.90C2
It was observed from the equation and perturbation plot (Figure 2) that the amount of Span 60 and Lipoid S 100 had negative effect on encapsulation while amount of cholesterol had positive effect indicating that increase in concentration of cholesterol encapsulate more amount of drug. It may be due to the fact that cholesterol forms hydrogen bond with non ionic surfactant and impart stability to niosomes and hence, more amount of drug is encapsulated18. It is reported that Span 60 has HLB value of 4.7 and decreased HLB value is also responsible for decreases encapsulation efficiency than that of surfactant with high HLB19.
Table 3: Results of responses measured for 20 CCD batches
| Experiment | Y1 (% EE) (Mean±SD) | Y2 (% CDR) (Mean±SD) |
|---|---|---|
| CD 1 | 80.85±0.19 | 74.8±0.94 |
| CD 2 | 89.31±0.32 | 84.17±0.65 |
| CD 3 | 80.18±0.31 | 71.86±0.43 |
| CD 4 | 76.89±0.12 | 95.4±0.64 |
| CD 5 | 84.75±0.24 | 90.53±0.84 |
| CD 6 | 81.03±0.34 | 90.9±0.89 |
| CD 7 | 84.94±0.16 | 85.47±0.51 |
| CD 8 | 75.46±0.18 | 96.06±0.14 |
| CD 9 | 82.42±0.24 | 85.01±0.72 |
| CD 10 | 74.36±0.22 | 97.16±0.69 |
| CD 11 | 84.59±0.15 | 90.9±0.5 |
| CD 12 | 76.48±0.28 | 95.63±0.94 |
| CD 13 | 76.01±0.37 | 82.1±0.63 |
| CD 14 | 90.85±0.26 | 80±0.66 |
| CD 15 | 81.2±0.23 | 87.3±0.35 |
| CD 16 | 81.37±0.27 | 86.76±0.83 |
| CD 17 | 81.76±0.33 | 85.3±0.45 |
| CD 18 | 81.23±0.28 | 84.15±0.81 |
| CD 19 | 81.66±0.35 | 86.13±0.34 |
| CD 20 | 81.46±0.16 | 87.46±0.81 |
| Optimized | 83.74±0.72 | 86.62±0.85 |
Effect of composition variable on % drug release
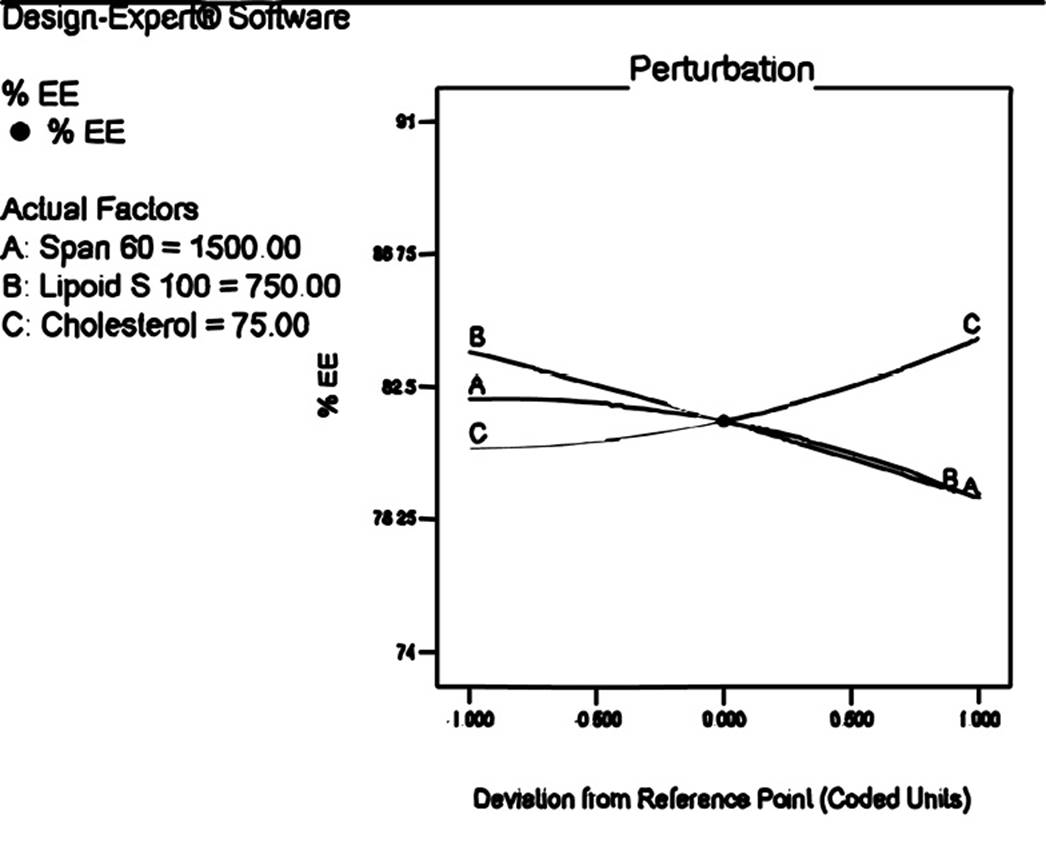
The percentage cumulative drug release was observed in the range of 71.86±0.43 to 97.16±0.69 (Table 3). Following is the polynomial equation for %CDR:
% CDR= +86.26+4.71*A+1.20*B+2.43*C+3.05*A*B-2.74*A*C-1.02*B*C+1.21*A2+1.99*B2-2.33*C2
The component of formulation; Span 60, Lipoid S 100 and cholesterol had positive coefficients. The perturbation plot (Figure 3) reveled that curvature of factor A i.e. Span 60 demonstrated significant proportional effect on drug release as compare to Lipoid S 100 and cholesterol. It was observed from the release profile that the vesicular system prolonged the release of etodolac for 24 hrs. The release studies have demonstrated presence of some amount of drug may bind to surface because of that initial burst release of etodolac followed by slow release was observed. The prolonged release for longer period of time could improve patient compliance as it reduces application frequency20.
Statistical evaluation suggested that quadratic model was significant. The release mechanism was studied for all formulation and it was observed that it followed zero order release kinetic. The comparative release profiling of optimized formulation and marketed formulation is given in Figure 4.
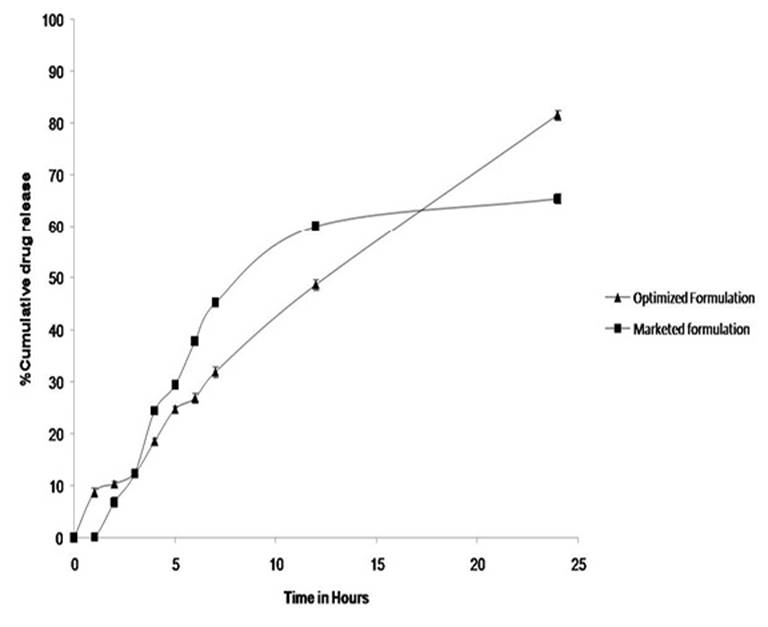
In order to optimize the formulation desirability approach test was used where the goal for the entrapment efficiency was set to 75% to 80% and maximum for %CDR. From the 20 solutions obtained, the one solution which has desirability equal to 1 was selected and experiment was conducted in triplicate and evaluated for %EE and %CDR. The predicted and observed responses of optimized formulation are shown in Table 4.
Table 4: Predicted and observed responses for optimized formulation
| Span 60 (mg) | 1800 | Response | Predicted | Observed |
|---|---|---|---|---|
| Lipoid S 100 (mg) | 971 | EE (%) | 75.06 | 74.12 |
| Cholesterol (mg) | 58 | CDR (%) | 97.22 | 95.08 |
Regression analysis of the data is presented in Table 5.
Evaluation of proniosomal gel
pH and viscosity
This finding is expected since, topical systems are directly applied on the skin; they should not causes skin irritation or disruption of the skin structure. The pH of all formulation batches was in between 4.2±0.20 to 5.9±0.1 and optimized formulation had 5.6±0.05. Therefore, it was reasonable to expect that formulations were found to be compatible with skin pH. The viscosity was related to amount of Span 60. Batches containing lower batches of Span 60 had low viscosity. The viscosity of optimized formulation was 6686±147.14 cPs which is what one could expect from the gel. All the formulations had shown shear thinning properties.
Zeta potential and vesicle size
The zeta potential demonstrates the potential of the parameter for the stability of the colloidal dispersions like vesicular formulation. Higher is the value of zeta potential; greater is the repulsion because of reduced aggregation between the particles18,19. The zeta potential of optimized formulation was -19.4mV; the negative value was due to negatively charged phospholipids present in soy lecithin. Because of negative charge; the droplets were repelled away from each other; avoid aggregation and impart stability. Another important parameter is size of the vesicle. The average vesicle size of the optimized formulation was found to be 211.9 nm with PDI of 0.58. This revealed that the vesicles had larger size and were relatively homogenous. The larger size of the vesicles containing soy lecithin is due to the difference in the composition of soy and egg derived lecithin20.
The rate of spontaneity i.e. formation of vesicles per 1 cubic mm was ranged from 51.25±0.66 to 141.75±0.62. The optimized formulation had the rate of 94.75±0.54.
Ex-vivo drug release
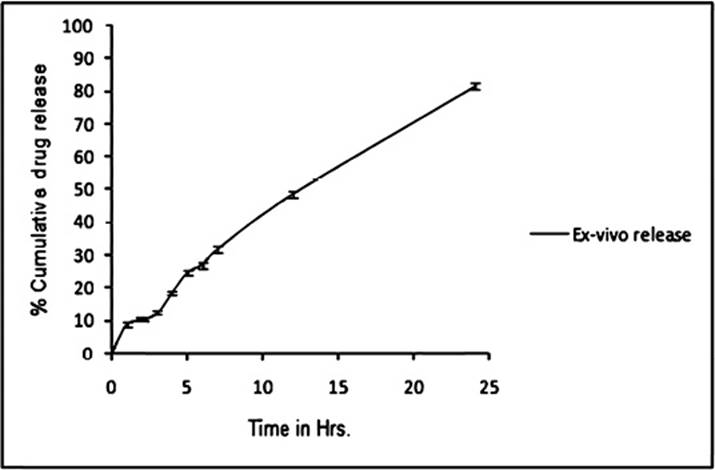
The formulations were studied using rat's excised skin and it was observed that the release of drug was more than that of marketed gel. Release behaviour justified the significance of vesicular systems. It seems that proniosomal formulation can serve as a penetration enhancer by modifying the lipids and increasing the fluidity which leads to weakening of skin's barrier properties21. The release profile is shown in Figure 5.
SEM study
The SEM analysis enables the determination of surface morphologies like shape and size of proniosomes than its hydrated form i.e. niosomes. As illustrated in Figure 6; the photomicrographs indicated that the vesicles had size around 200 nm and spherical in shape. The increased size of the vesicle was not related to instability issues but may be due to removal of moisture during SEM analysis from the preparation causing them to aggregate.

Anti-inflammatory activity
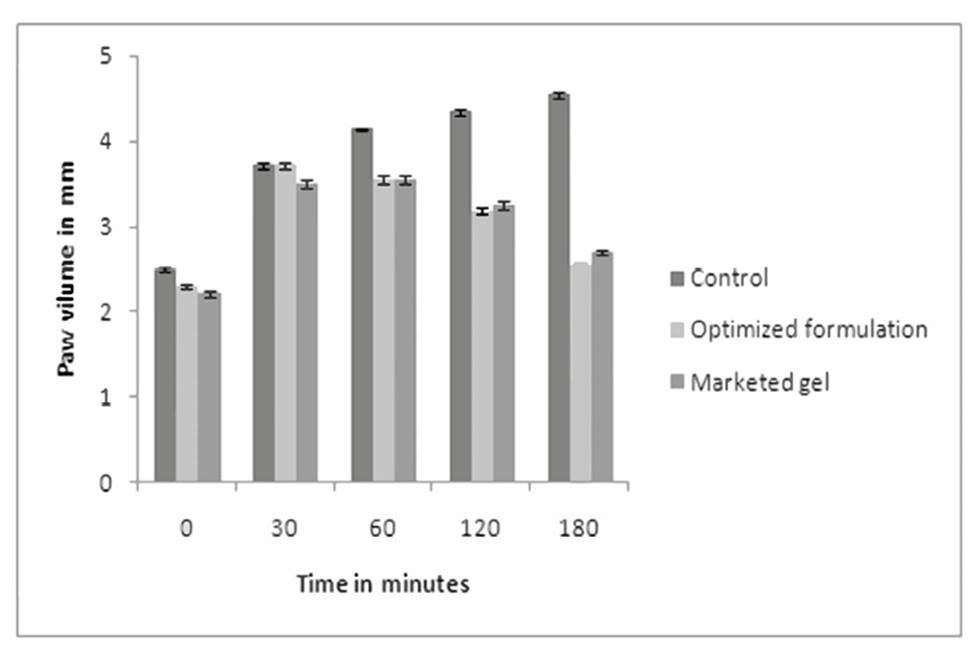
The paw volume of control, standard and test formulation treated animals was measured. The % inhibition of paw volume was compared amongst standard and test groups. It was observed that the % inhibition by marketed gel and proniosomal gel was 43.61 and 40.52 % respectively which indicates that formulated proniosomal gel was as effective as standard gel in its anti-inflammatory activity. The results are presented in Figure 7. The P value is<0.0294 is considered as extremely significant by applying ANOVA test followed by Dunnett test.
Conclusion
The work has demonstrated the use of Span 60, Lipoid S 100 (soy lecithin) and cholesterol appropriate for the development of etodolac proniosomal gel. The proniosomal gel formulation was stable for the period of 3 months. It had proper features for transdermal delivery. The ex vivo study indicated more release than marketed formulation; it could be due to increased penetration ability of the vesicular system. The anti-inflammatory studies showed that the formulation was comparable with marketed one. Thus, the developed topical proniosomal formulation may prove to be a promising carrier for other anti-inflammatory drugs too in the context of simple production and scale up.

