










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versão impressa ISSN 1130-0108
Rev. esp. enferm. dig. vol.101 no.12 Madrid Dez. 2009
Peutz-Jeghers syndrome and duodeno-jejunal adenocarcinoma - therapeutic implications
Síndrome de Peutz-Jeghers y adenocarcinoma duodenal-yeyunal: implicaciones terapéuticas
J. A. Cienfuegos, J. Baixauli, G. Zozaya, A. Bueno, J. Arredondo, F. M. Regueira, R. Angós1, J. L. Hernández-Lizoáin and M. A. Idoate2
Departments of General Surgery, 1Digestive Diseases, and 2Pathology. Clínica Universidad de Navarra. Pamplona, Spain
ABSTRACT
The Peutz-Jeghers syndrome (PJS) is an autosomal dominant hamartomatous poliposis describred in 1921. Hemminki in 1997 described the presence of LKB-1 mutation tumor-suppressor gen.
The patients with PJS develop a higher cumulative incidence of gastrointestinal, pancreas and extraintestinal tumors, being occasion of a renew interest on hamartomatous polyposis syndromes regarding the clinical care, cancer surveillance treatment and long term follow-up.
We report the case of a 38 years old male, diagnosed of PJS who developed a multiple adenocarcinoma in duodenum and yeyunum. Surgically treated and with a long-term free disease survival of 11 years represents the sixth case reported in the spanish literature of PJS associated with a gastrointestinal tumor.
A critical review, molecular alterations and the established criteria of tumor screening and surveillance are reviewed.
Key words: Peutz-Jeghers syndrome. Hamartomatous. Carcinogenesis. Intestinal polyposis. Hereditary cancer.
Introduction
Peutz-Jeghers syndrome (PJS) is an autosomal dominant disease characterized by the presence of multiple hamartomatous polyps throughout the entire gastrointestinal tract and mucocutaneous melanin pigmentation (1-6).
Hemminki, in 1997, described a deletion in the short arm of chromosome 19, a mutation in the LKB-1 gene (also known as STK11) (7-10). In this syndrome a high cumulative incidence of tumors in the gastrointestinal tract and other locations (breast, lung, gynecologic) has been reported. A renewed interest in its molecular pathogenesis and in the surveillance and screening strategy has been observed (1,4-6).
Case report
A 38-year-old male with a previous diagnosis of PJS was hospitalized in another center for nausea, vomiting, and abdominal pain. Upper endoscopy revealed a complete obstruction of the duodenal lumen by an extrinsic mass, which prompted referral to our center.
An abdominal computed tomography (CT) scan (Fig. 1) revealed a solid mass with hypodense areas and infiltrating appearance compromising the duodenal lumen, and a jejuno-jejunal intussusception was described. Gastroscopy confirmed the presence of multiple gastric polyps and complete duodenal obstruction. Citology revealed malignant cells.
The patient underwent pancreticoduodenectomy with a simultaneous resection of 23 cm of jejunum with signs of intussusception. The patient required no blood transfusion and had an uneventful postoperative course except for an episode of self-limited hematochezia.
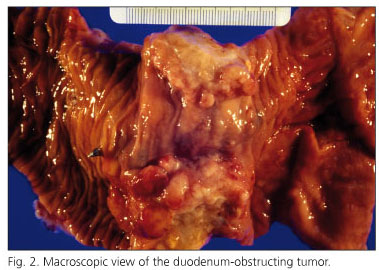
The histopathological examination revealed what follows: "The main tumor measured 6 cm in diameter and was located in the duodenum. This tumor obstructed the duodenum, infiltrated the pancreas, and showed large cystic areas between 5 and 9 mm in diameter (Fig. 2). The duodenal tumor showed an atypical epithelial glandular growth pattern, which infiltrated the wall of the small bowel and pancreas (Fig. 3). Two other similar infiltrating tumors, which measured between 1.5 and 5 cm in diameter, and occupied the entire intestinal wall, were located in the jejunum. The atypical glands were frequently cystic and showed characteristics similar to those in the duodenum. No metastatic tumor was identified in the periportal or peripancreatic lymph nodes. Primary well-differentiated multifocal adenocarcinomas originating from pre-existing polyps were diagnosed.

Multiple polyps in the stomach and jejunum were observed. Gastric polyps were numerous and measured between 1 and 4 mm in diameter. Three pediculated jejunal polyps, which measured up to 3 cm in diameter, were observed. Histologically, these tumors showed either hyperplastic or adenomatous characteristics. The glands were cystic and covered with moderately to severely dysplastic mucosecretory epithelium. Frequent bundles of smooth muscle from the muscularis mucosae were spread among the atypical glands".
A PJS diagnosis had been first established in 1989 when the patient was 29 years old. The patient required and urgent intestinal resection for an intestinal intussusception and subsequent bowel obstruction. Several hamartomatous polyps and phenotypical features - mucocutaneous melanosis - were assessed. There was no family history of "intestinal problems".
The patient has been followed up for 11 years and remains asymptomatic and recurrence-free. A surveillance program was implemented with gastroscopies, double-balloon enteroscopies, two capsule endoscopy studies, and seven colonoscopies. Fifteen gastric hyperplastic polyps and 11 hamartomatous polyps, 9 jejunal hamartomatous polyps without epithelial dysplasia, 7 colon hamartomatous polyps, and 3 adenomatous polyps with low-grade dysplasia foci were removed without complications.
Discussion
Peutz-Jeghers syndrome (PJS) was first described by Peutz en 1921, and then by Jeghers in 1949 (11,12). In 1954 Bruwer "coined" the eponym "Peutz-Jeghers" (13). PJS is characterized by the presence of multiple adenomatous and hamartomatous polyps along the gastrointestinal tract in association with melanocytic pigmentation around the mouth, but also in the hands and feet and axillary pits. An incidence of 1/8,300 to 1/200,000 live births has been estimated, and the condition is an autosomal dominant disease that affects both males and females, and is found in all racial groups.
Three series reviewed 404 PJS patients and reported jejunal polyps in 78%, colon polyps in 42%, gastric polyps in 38%, and rectal polyps in 28% of patients (1-3,6). Epithelial dysplasia foci and invasive carcinoma have been reported in 26% of polyps (9).
Hemminki was first to describe a deletion in the short arm of a chromosome 19 region, which was later identified as a mutation in the LKB-1 gene (19p 13.3), also known as STK11 (serine/threonine-protein/kinase 11). LKB-1 acts as a tumor suppressor gene and is believed to regulate cell orientation, polarization and apoptosis. A loss of LKB-1 function leads to hamartomatosus polyp formation and tumor formation because of epithelial polarity disruption. This molecular alteration has been reported in 30 to 82% of PJS patients (8-10,14-20).
In addition to the clinical diagnosis and polyp-related complications such as intestinal obstruction (14%), abdominal pain (23%), rectal bleeding (14%), and polyp intussusception (40%) (1-3,6,10), the most challenging clinical aspect is early tumor or preinvasive lesion identification.
Germ-line mutations are present in 70% of families with complete penetrance. The cumulative risk of developing cancer is 93% at 65 years of age, with a median age at presentation of 43 years. There have been no differences in cancer incidence between patients with and without molecular mutations.
The reported case shows a strong correlation with the natural course of the disease. The patient was diagnosed at 29 years of age on presenting with an acute abdominal complication (1-3,6,10,21,22) and developed multiple gastrointestinal cancer at 38 years. It represents the sixth case of intestinal adenocarcinoma in PJS in the Spanish literature, with previous cases being gastrointestinal adenocarcinomas (stomach, jejunum, jejunum-ileum, and colon) and one case of simultaneous renal, rectal and cholangiocarcinoma (23-27).
Dozois et al reported in 1969 eleven cases of gastrointestinal cancer out of 321 PJS patients (4 in the stomach, 3 the in duodenum, 1 in the ileum, and 3 in the colon and rectum) (28).
In 2000 Gianderlo conducted a meta-analysis in order to define cancer risk in a PJS population, and studied 210 patients from 6 case-series published in Holland and the UK. The relative risk of cancer was 15.2 with no gender-specific differences. Age at presentation was 42.9 ± 10.2 years, similar to our reported case (38 years). The cumulative risk of cancer was 93% at 65 years. The highest cumulative incidence was: breast (54%), colon (39%), pancreas (36%), stomach (29%), and ovary (21%) (29).
In a later report of 419 PJS patients the cumulative risk of cancer at age 70 was 85%, four times superior to the general population. Again colo-rectal and breast tumors were most frequent (30). The relative risk of gastric and small-bowel cancer was 84; 213 and 520, respectively, when compared to the general population (95% CI) (29).
In Gianderlo's study, six patients developed pancreatic cancer; age at diagnosis was 41 years with a relative risk of 36% for this tumor, which supports the need for surveillance to facilitate early detection for pancreatic cancer (31-33).
Hearle reported 11 cases of pancreatic cancer in 4,109 individuals with PJS syndrome, but did not report the outcomes of these cases (30).
Today the existence of precursor lesions giving rise to invasive pancreatic cancer is well established, with a pattern similar to the adenoma-carcinoma sequence previously described for colon cancer (34-36). This offers an opportunity to detect local precursor lesions such as pancreatic intraepithelial neoplasia (Pan IN) or early invasive lesions (< 3 cm), which are amenable to curative resection. Screening for invasive pancreatic cancer and its precursors is currently recommended with endoscopic ultrasonography (EUS) and computed tomography (CT) annually, followed by endoscopic retrograde cholangiopancreatography (ERCP) and fine-needle aspiration for suspect lesions (37).
In the Johns Hopkins experience seven patients out of 38 PJS patients underwent pancreatoduodenectomy. One of them had an adenocarcinoma of 2.8 cm, and remains free of disease five years after surgery. The other six had pain with various grades (38).
PJS patients must be included in specific follow-up programs with screening for pancreas cancer (39-41).
Table I depicts the most numerous series and the incidence of cancer in PJS patients, and table II shows screening techniques and surveillance strategies (49-54).


References
1. Hemminki, A. The molecular basis and clinical aspects of Peutz-Jeghers syndrome. Cell Mol Life Sci 1999; 55: 735-50. [ Links ]
2. Utsunomiya J, Gocho H, Miyanaga T, Hamaguchi E, Kashimure A. Peutz-Jeghers syndrome: its natural course and management. Johns Hopkins Med J 1975; 136: 71-82. [ Links ]
3. Bartholomew, L.G.; Dahlin, D.C.; Waugh, J.M. Intestinal polyposis associated with mucocutaneous melanin pigmentation (Peutz-Jeghers syndrome). Proc Staff Meet Mayo Clin 1957; 32: 675-80. [ Links ]
4. McGarrity TJ, Amos C. Peutz-Jeghers syndrome: clinicopathology and molecular alterations. Cell Mol Life Sci 2006; 63: 2135-44. [ Links ]
5. Mehenni H, Resta N, Guanti G, Mota-Vieira L, Lerner A, Peyman M, et al. Molecular and clinical characteristics in 46 families affected with Peutz-Jeghers syndrome. Dig Dis Sci 2007; 52: 1924-33. [ Links ]
6. Schreibman IR, Baker M, Amos C, McGarrity TJ. The hamartomatous polyposis syndromes: a clinical and molecular review. Am J Gastroenterol 2005; 100: 476-90. [ Links ]
7. Hemminki A, Tomlinson I, Markie D, Järvinen H, Sistonen P, Björkqvist AM, et al. Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hybridization and targeted linkage analysis. Nat Genet 1997; 15: 87-90. [ Links ]
8. Yoo LI, Chung DC, Yuan J. LKB1 -a master tumour suppressor of the small intestine and beyond. Nat Rev Cancer 2002; 2: 529-35. [ Links ]
9. Yantiss RK, Antonioli DA. Polyps of the small intestine. En: Odze RD, Goldblum JR, editors. Surgical pathology of the GI tract, liver, biliary tract and pancreas. 2nd ed. Philadelphia: Saunders Elservier; 2009. p. 447-80. [ Links ]
10. Calva D, Howe JR. Hamartomatous polyposis syndromes. Surg Clin N Am 2008; 88: 779-817. [ Links ]
11. Peutz J. A very remarkable case of familial polyposis of mucous membrane of intestinal tract and accompanied by peculiar pigmentations of skin and mucous membrane. Ned Tijdschr Geneskd 1921; 10: 134-46. [ Links ]
12. Jeghers H, McKusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits; a syndrome of diagnostic significance. N Engl J Med 1949; 241: 1031-6. [ Links ]
13. Bruwer A, Bargen JA, Kierland RR. Surface pigmentation and generalized intestinal polyposis (Peutz-Jeghers syndrome). Proc Staff Meet Mayo Clin 1954; 29: 168-71. [ Links ]
14. Van der Flier LG, Clevers H. Stem cells, self-renewal and differentiation in the intestinal epithelium. Ann Rev Physiol 2009; 71: 241-60. [ Links ]
15. Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998; 391: 184-7. [ Links ]
16. Jenne DE, Reimann H, Nezu J, Friedel W, Loff S, Jeschke R, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat Genet 1998; 18: 38-43. [ Links ]
17. Aaltonen LA. Peutz-Jeghers syndrome. En: Vogelstein B, Kinzer KW, editors. The genetics basis of human cancer. McGraw Hill Companies; 2002. p. 337-41. [ Links ]
18. Martin SG, St Johnston D. A role for Drosophila LKB1 in anterior-posterior axis formation and epithelial polarity. Nature 2003; 421: 379-84. [ Links ]
19. Karuman P, Gozani O, Odze RD, Zhou XC, Zhu H, Shaw R, et al. The Peutz-Jegher gene product LKB1 is a mediator of p53-dependent cell death. Mol Cell 2001; 7: 1307-19. [ Links ]
20. Yoo LI, Chung DC, Yuan J. LKB1--a master tumour suppressor of the small intestine and beyond. Nat Rev Cancer 2002; 2: 529-35. [ Links ]
21. González Muñoz JL, Angoso Clavijo M, Esteban Velasco C, Rodríguez Pérez A, Muñoz Bellvis L, Gómez Alonso A. Diagnóstico de síndrome de Peutz-Jeghers. Rev Esp Enferm Dig 2007; 99: 167. [ Links ]
22. Gutiérrez Benjumea A, Rojo García J, Aguilera Llovet MA, García Arqueza C, Casanovas Lax J, Aguayo Maldonado J. Síndrome de Peutz-Jeghers. An Esp Pediatr 2001; 55: 161-4. [ Links ]
23. Bujanda L, Beguiristain A, Villar JM, Cosme A, Castiella A, Arriola JA, et al. Adenocarcinoma gástrico sobre pólipo hamartomatoso en el síndrome de Peutz-Jeghers. Gastroenterol Hepatol 1996; 19: 452-5. [ Links ]
24. Rodríguez JM, Picardo A, Torres AJ, García Calvo M, Ortega L, Martínez S, et al. Síndrome de Peutz-Jeghers con malignización de un pólipo hamartomatoso. Rev Esp Enferm Dig 1993; 84: 56-9. [ Links ]
25. Fernández-Llamazares J, Piñol M, Ojanguren I, Armengol M, Oller B, Salvá A. Hamartoma degenerado en un síndrome de Peutz-Jeghers. Caso Clínico. Cir Esp 1988; 44: 511-4. [ Links ]
26. Hidalgo L, Villanueva A, Soler T, Matías Guiu X, Capellá G. Alteraciones moleculares en adenocarcinoma de intestino delgado asociado a síndrome de Peutz-Jeghers. Rev Esp Enferm Dig 1996; 88: 137-40. [ Links ]
27. Cosme A, Ojeda E, San Vicente MT, Barrio J, Bujanda L, López P. Síndrome de Peutz-Jeghers asociado a múltiples tumores epiteliales. Gastroenterol Hepatol 2001; 24: 495-9. [ Links ]
28. Dozois RR, Judd ES, Dahlin DC, Bartholomew LG. The Peutz-Jeghers syndrome. Is there a predisposition to the development of intestinal malignancy? Arch Surg 1969; 98: 509-17. [ Links ]
29. Giardiello FM, Brensinger JD, Tersmette AC, Goodman SN, Petersen GM, Booker SV, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000; 119: 1447-53. [ Links ]
30. Hearle N, Schumacher V, Menko FH, Olschwang S, Boardman LA, Gille JJ, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006; 12: 3209-15. [ Links ]
31. Greenhalf W, Grocock C, Harcus M, Neoptolemos J. Screening of high-risk families for pancreatic cancer. Pancreatology 2009; 9: 215-22. [ Links ]
32. Su GH, Hruban RH, Bansal RK, Bova GS, Tang DJ, Shekher MC, et al. Germline and somatic mutations of the STK11/LKB1 Peutz-Jeghers gene in pancreatic and biliary cancers. Am J Pathol 1999; 154: 1835-40. [ Links ]
33. Lochan R, Daly AK, Reeves HL, Charnley RM. Genetic susceptibility in pancreatic ductal adenocarcinoma. Br J Surg 2008; 95: 22-32. [ Links ]
34. Singh M, Maitra A. Precursor lesions of pancreatic cancer: molecular pathology and clinical implications. Pancreatology 2007; 7: 9-19. [ Links ]
35. Maitra A, Hruban RH. Pancreatic cancer. Annu Rev Pathol 2008; 3: 157-88. [ Links ]
36. Maitra A, Fukushima N, Takaori K, Hruban RH. Precursors to invasive pancreatic cancer. Adv Anat Pathol 2005; 12: 81-91. [ Links ]
37. Hruban RH, Takaori K, Klimstra DS, Adsay NV, Albores-Saavedra J, Biankin AV, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol 2004; 28: 977-87. [ Links ]
38. Canto MI, Goggins M, Yeo CJ, Griffin C, Axilbund JE, Brune K, et al. Screening for pancreatic neoplasia in high-risk individuals: an EUS-based approach. Clin Gastroenterol Hepatol 2004; 2: 606-21. [ Links ]
39. Giardiello FM, Trimbath JD. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepatol. 2006; 4: 408-15. [ Links ]
40. Latchford A, Greenhalf W, Vitone LJ, Neoptolemos JP, Lancaster GA, Phillips RK. Peutz-Jeghers syndrome and screening for pancreatic cancer. Br J Surg 2006; 93: 1446-55. [ Links ]
41. Boardman LA, Thibodeau SN, Schaid DJ, Lindor NM, McDonnell SK, Burgart LJ, et al. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med 1998 1; 128: 896-9. [ Links ]
42. Utsunomiya J, Nakamura T. The occult osteomatous changes in the mandible in patients with familial polyposis coli. Br J Surg 1975; 62: 45-51. [ Links ]
43. Linos DA, Dozois RR, Dahlin DC, Bartholomew LG. Does Peutz-Jeghers syndrome predispose to gastrointestinal malignancy? A later look. Arch Surg 1981; 116: 1182-4. [ Links ]
44. Giardiello FM, Welsh SB, Hamilton SR, Offerhaus GJ, Gittelsohn AM, Booker SV, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med 1987; 316: 1511-4. [ Links ]
45. Spigelman AD, Murday V, Phillips RK. Cancer and the Peutz-Jeghers syndrome. Gut 1989; 30: 1588-90. [ Links ]
46. Boardman LA, Thibodeau SN, Schaid DJ, Lindor NM, McDonnell SK, Burgart LJ, et al. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med 1998; 128: 896-9. [ Links ]
47. Choi HS, Park YJ, Youk EG, Yoon KA, Ku JL, Kim NK, et al. Clinical characteristics of Peutz-Jeghers syndrome in Korean polyposis patients. Int J Colorectal Dis 2000; 15: 35-8. [ Links ]
48. Lim W, Olschwang S, Keller JJ, Westerman AM, Menko FH, Boardman LA, et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology 2004; 126: 1788-94. [ Links ]
49. Learn PA, Kahlenberg MS. Hereditary colorectal cancer syndromes and the role of the surgical oncologist. Surg Oncol Clin N Am 2009; 18: 121-44. [ Links ]
50. Gammon A, Jasperson K, Kohlmann W, Burt RW. Hamartomatous polyposis syndromes. Best Pract Res Clin Gastroenterol 2009; 23: 219-31. [ Links ]
51. Burke CA, Santisi J, Church J, Levinthal G. The utility of capsule endoscopy small bowel surveillance in patients with polyposis. Am J Gastroenterol 2005; 100: 1498-502. [ Links ]
52. Soares J, Lopes L, Vilas Boas G, Pinho C. Wireless capsule endoscopy for evaluation of phenotypic expression of small-bowel polyps in patients with Peutz-Jeghers syndrome and in symptomatic first-degree relatives. Endoscopy 2004; 36: 1060-6. [ Links ]
53. Pennazio M, Rondonotti E, de Franchis R. Capsule endoscopy in neoplastic diseases. World J. Gastroenterol 2008; 14: 5245-53. [ Links ]
54. Pérez-Cuadrado E, Más P, Hallal H, Shanabo J, Muñoz E, Ortega I, et al. Enteroscopia de doble balón: estudio descriptivo de 50 exploraciones. Rev Esp Enferm Dig 2006; 98: 73-81. [ Links ]
![]() Correspondence:
Correspondence:
Javier A. Cienfuegos.
Departamento de Cirugía General.
Clínica Universidad de Navarra.
Avda. Pío XII, 36. 31008 Pamplona.
e-mail: fjacien@unav.es
Received: 02-07-09.
Accepted: 15-07-09.

