










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkSr. Director:
Presentamos el caso de una mujer de 44 años sin antecedentes, que presenta cuadro febril de 38,7°C de predominio vespertino, sin foco, escalofríos ni tiritona, de 2 semanas de evolución; es tratado como cuadro seudogripal con AINE y antitérmicos. Posteriormente desarrolla exantema cutáneo generalizado no pruriginoso por lo que acude a urgencias, donde se objetiva anemia (11,7 g/dl hb) y leucopenia leves con ligera linfopenia (3.640 leucos/ul, 18% linfos), PCR 22 e hipertransaminemia leve. Función renal normal (creatinina 0,73 mg/dl, orina simple normal), exploración anodina salvo por una temperatura de 37,7°C y exantema confluente que incluye palmas/plantas y blanquea tras vitropresión. Radiografía de tórax normal. Con el diagnóstico de síndrome febril exantemático con alteración hepática y hematológica ingresa para estudio, pautándose doxiciclina.
En días posteriores se detecta hepatoesplenomegalia y derrame pericárdico, VSG 110 y frotis sin atipias celulares pero células LUC 11%. Hierro, fólico, B12, IgA, IgG, C3-4, ANCA, ASLO, péptido citrulínico normales. Proteinograma: elevación policlonal de la fracción gamma, IgM elevada, FR 300 UI/ml y crioglobulinas negativas. ANA 1/80, DNA, ENA negativos salvo U1-RNP 24 UI/ml. Serología: VHB, VHC, VIH, Legionella, CMV, parvovirus B19, Leptospira, C. burneti, R. coronii, neumococo, hemocultivos negativos; VEB IgM dudoso/IgG positivo.
Permanece febril, presentando en el quinto día de ingreso intensificación de la fiebre con escalofríos/tiritona e insuficiencia respiratoria, detectándose atelectasia/condensación bibasal con derrame pleural simétrico (fig. 1A) y procalcitonina 4,39 ng/ml, recibiendo antibioterapia de amplio espectro.
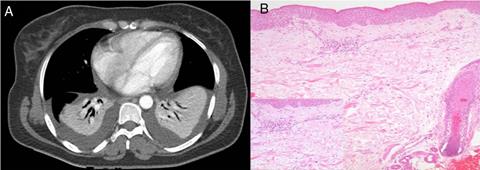
Figura 1 TAC (A): atelectasia/condensación bibasal con derrame simétrico, derrame pericárdico. Biopsia piel (B): espongiosis epidérmica intracelular focal, ligero edema y extravasación hemática, y vasos dérmicos ocupados por células inflamatorias (aumento: linfocitos y polimorfonucleares con aislados eosinófilos).
Nefrológicamente, proteinuria 1,8 g/día, sedimento 40 H/C (28% dismorfias), colesterol normal, albúmina 2,2; mínimo edema pretibial simétrico, persistiendo la presión arterial y la creatinina normales, así como la ecografía. No presentaba ninguna otra clínica sistémica. La biopsia dérmica confirma proceso urticarial (fig. 1B). Se realiza entonces biopsia renal que informa de una glomerulonefritis proliferativa mesangial con depósitos inmunes predominantes IgM/C1q. Se recibe PCR VEB+ en sangre, siendo negativa la hibridación in situ en riñón (fig. 2). Se diagnostica de mononucleosis infecciosa (MI) con afectación hematológica, pleuropericárdica, hepática y renal con glomerulonefritis proliferativa mesangial por inmunocomplejos. Evoluciona favorablemente, quedando asintomática y con negativización de la PCR-VEB. Al año del cuadro mantiene remisión, aunque persiste anti-U1-RNP+ y artralgias generalizadas tratadas con analgesia habitual que ya presentaba desde hacía años. No ha positivizado ningún otro autoanticuerpo ni presentado ninguna otra clínica sistémica.
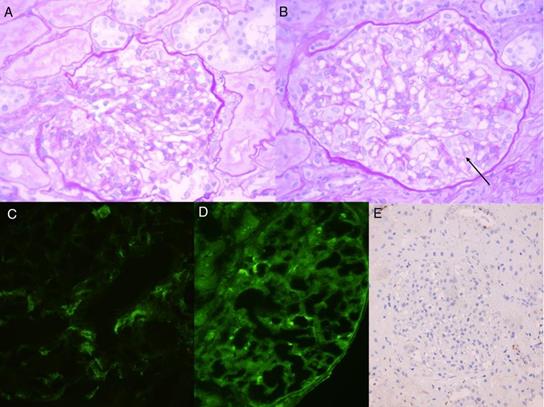
Figura 2 Biopsia renal. PAS 60x: proliferación mesangial (A), podocitos prominentes (B, flecha). IF: IgM mesangial difuso y global (C), C1q mesangial segmentario (D). Hibridación in situ para VEB negativa (EBER, E).
La MI en adultos se presenta con frecuencia de forma atípica1, en ausencia de faringoamigdalitis, adenopatías y linfocitosis/atipias linfocíticas. El derrame pleural o pericárdico son excepcionales, describiéndose tan solo aislados casos en la literatura2. Todo ello, junto a la leucopenia-anemia, serositis y positividad de U1-RNP, hizo pensar en la coexistencia de infección y autoinmunidad.
La prevalencia de afectación renal en la MI no está bien filiada dado que suele pasar desapercibida con tan solo alteraciones urinarias asintomáticas (14-17%)3. Los casos floridos publicados son aislados y fundamentalmente de tipo tubulointersticial; le sigue una minoría de glomerulopatías varias, mediadas o no por inmunocomplejos, que pueden presentarse en solitario o coexistiendo con la afectación intersticial4. La presencia de insuficiencia renal se estima entre 1,6-4,8%3. En cuanto a su patogénesis, se han barajado varios mecanismos no excluyentes: daño por inmunocomplejos o daño tóxico-citopático directo, más encontrado este último en las lesiones intersticiales, en las que se ha aislado el virus y encontrado supremacía de las células CD8+. En nuestro caso no logramos encontrar el virus mediante hibridación in situ y apenas se describe daño tubulointersticial, por lo que deducimos que el daño fue mediado por anticuerpos/inmunocomplejos más que por citotoxicidad directa. Los depósitos encontrados fueron mayoritariamente IgM/C1q. Andres et al.5 describen un caso similar con depósitos mesangiales IgM/C3, siendo esta la misma IgM sérica heterófila contra antígeno Paul Bunnell. Esto es porque la respuesta inmune humoral frente al EBV es principalmente a expensas de IgM6. Por otra parte, se ha descrito activación de las 2 vías del complemento en la MI7.
El porqué el VEB hiperestimula en algunos casos la inmunidad del huésped se desconoce, pero se ha detectado in vitro e in vivo que la IgM natural producida por la activación policlonal de los linfocitos B durante la MI tiene propiedad antihistona8. Niller et al.9 relacionaron diferentes antígenos del VEB con la activación de los linfocitos B autorreactivos en la esclerosis múltiple, LES y AR. A ese respecto, la positividad para U1-RNP también la atribuimos a la activación por parte del virus de linfocitos B secretores de inmunoglobulinas específicas contra dicha proteína, por reacción cruzada con antígenos del propio VEB10. Desconocemos por qué, a pesar de presentar ese anticuerpo, no desarrolla clínica asociada al mismo y su patogenicidad en un futuro, si bien el título que ha presentado nunca ha sido muy elevado. Se ha realizado HLA de la paciente sin encontrarse alelos predisponentes para enfermedades autoinmunes.
En cuanto al tratamiento, la glomerulopatía remitió espontánea y paralelamente al cuadro mononucleósico. En la literatura, algunos casos han recibido corticoterapia, sobre todo las tubulointersticiales, no en nuestro caso por la leve afectación clinicohistológica renal (si bien la hipercelularidad mesangial era difusa en algunos glomérulos, era segmentaria en otros y en general de cuantía leve). Tampoco aciclovir, por la escasa eficacia del mismo en la literatura.
Concluimos que la frecuente presentación atípica de la MI en el adulto puede llevar a retardar el diagnóstico. La afectación renal suele ser tubulointersticial por citotoxicidad en la que se puede encontrar el virus tejido, y menos frecuentemente glomerular; a tener en cuenta la patogenia por inmunocomplejos y activación del complemento. El VEB es considerado un «trigger» de fenómenos autorreactivos que pueden perseverar en el tiempo y provocar, sobre un lecho predisponente, una enfermedad autoinmune.

