










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNutrición Hospitalaria
versión On-line ISSN 1699-5198versión impresa ISSN 0212-1611
Nutr. Hosp. vol.21 supl.3 Madrid may. 2006
ARTÍCULO
Fisiopatología de la caquexia neoplásica
Pathophysiology of neoplasic cachexia
J. M. Argilés, S. Busquets, F. J. López-Soriano y M. Figueras
Departament de Bioquímica i Biologia Molecular. Universitat de Barcelona. Spain.
Dirección para correspondencia
RESUMEN
La regulación del apetito y de los patrones alimenticios está mediada por diferentes factores psicológicos, gastrointestinales, metabólicos y nutricionales, así como por distintos mecanismos neuronales y endocrinos. El paciente canceroso anoréxico experimenta una sensación precoz de saciedad y una disminución del apetito. En algunas ocasiones, las causas de esta anorexia pueden derivarse del propio tratamiento anticanceroso (quimioterapia, radioterapia o inmunoterapia), que pueden inducir náuseas y vómitos en diferentes grados. También pueden contribuir a la reducción de la ingesta las alteraciones en la percepción de la comida y causas psicológicas (depresión). En ocasiones, la anorexia puede atribuirse a un efecto directo del tumor, cuando éste se localiza en el hipotálamo o en el propio aparato digestivo. Sin embargo, en la mayoría de los casos el origen de la anorexia asociada a caquexia parece ser las alteraciones metabólicas que sufre el paciente como consecuencia de la presencia del tumor.
Diferentes factores tanto de origen humoral y segregados por el huésped en respuesta al crecimiento tumoral, o bien segregados por las propias células tumorales, podrían jugar un papel importante en la respuesta anoréxica. Entre los primeros destaca el factor necrótico tumoral- α (TNF-α), una citoquina que parece ser la responsable de la mayor parte de las alteraciones metabólicas características de la caquexia cancerosa.
En definitiva, la anorexia parece ser más un efecto que la causa de la pérdida de peso y, de hecho, la disminución de la ingesta puede manifestarse después de que haya habido pérdida de peso. En cualquier caso, la malnutrición debida a una menor ingesta de alimentos no hace sino agravar el estado caquéctico, propiciando una especie de mecanismo de retroalimentación positivo que puede conducir finalmente a la muerte del paciente.
Palabras clave: Caquexia. Cáncer. Fisiopatología. Citoquinas. Proteólisis muscular.
ABSTRACT
The regulation of food intake is mediated by different psicological, gastrointestinal metabolic, nutritional and endocrine mechanisms. The cancer patient suffers from anorexia which results in early saciety and a reduction of appetite. Sometimes, the causes of the anorectic response are derived from the antitumoral treatment (chemotherapy, radiotherapy or immunotherapy), in some cases vomiting resulting in altered food intake. Alterations in the food taste and smell perception in addition to psychological dearrangements might also lead to the anorexia. Sometimes the tumour may play a direct effect when it is localised in either the hypothalamus or the digestive apparatus. However, in the majority of cases the origin of the anorexia associated with cancer cachexia seems to be due to the metabolic alterations induced by tumour burden.
Different factors of both humoral and tumoral origin play a role in cancer anorexia. For instance, tumour necrosis factor (TNF- ), a cytokine responsible for a great part of the metabolic alterations characteristic of cancer cachexia seems to be involved.
In conclusion, the cancer anorexia seems to be more an effect than the cause of the weight loss and in fact the decrease in food intake might take place after weight loss is evident. In any case, the malnutrition associated with a decrease of food intake worsens the cachectic state, favouring a kind of a positive feed-back mechanism that finally leads to the patient's death.
Key words: Cachexia. Cancer. Physiopathology. Cytokines. Muscle proteolysis.
Introducción: la pérdida de peso asociada al cáncer
La caquexia es un complejo síndrome que está presente en más de dos terceras partes del conjunto de pacientes que mueren de cáncer avanzado, pudiendo ser la causa directa de una cuarta parte de los fallecimientos por esta enfermedad. Asimismo, este síndrome también aparece asociado a otros estados patológicos, como pueden ser las infecciones crónicas o los traumatismos de distinta índole. La caquexia se caracteriza por una importante y progresiva pérdida de peso corporal, así como por anorexia, astenia, anemia, náuseas crónicas e inmunosupresión. De éstas, la pérdida de peso corporal es una de las más aparentes, y es atribuible principalmente a una disminución de la masamuscular y adiposa. La pérdida de masa muscular afecta no sólo al músculo esquelético, sino también al cardíaco, lo que puede ser el origen de disfunciones en este órgano, las cuales pueden llegar a representar más de un 20% de los fallecimientos asociados al cáncer.
A nivel clínico, la importancia de la caquexia es considerable por cuanto existe una clara correlación inversa entre el grado de caquexia y la supervivencia del paciente. Además, la caquexia implica siempre una prognosis desfavorable, una menor respuesta a la terapia (tanto cirugía como quimioterapia), y una disminución en la calidad de vida del paciente. En función del tipo de tumor, su incidencia puede variar entre un 20% y un 80%.
Los orígenes de la caquexia han de buscarse en dos aspectos fundamentales: una incrementada demanda calórica debida a la presencia del tumor (con la correspondiente competencia por los nutrientes entre las células del paciente y las del tumor), y la malnutrición debida a la anorexia (disminución en la ingesta) (fig. 1). Ello conlleva a la aparición de lo que ha sido denominado como "ayuno acelerado", con el consiguiente desarrollo de importantes cambios metabólicos en el paciente (tabla I). Dichos cambios están asociados a la presencia de diferentes factores circulantes, tanto humorales principalmente citoquinas) como de origen tumoral.


La anorexia es un componente casi universal de la caquexia. Resulta poco probable que la anorexia sea por sí sola responsable del desgaste que se observa en los pacientes cancerosos, aunque puede ser un factor que contribuya a dicho desgaste; además, el apetito y la capacidad para comer han sido descritos como los factores más importantes de la calidad de vida del paciente, tanto en el aspecto físico como en el psicológico. La anorexia parece ser más una consecuencia de la caquexia que una de sus causas, pues se puede desarrollar una vez la pérdida de peso ya ha aparecido, pero lo que sí parece establecerse es un ciclo de retroalimentación positiva entre el debilitamiento progresivo y la falta de apetito, que se agravan así mutuamente (fig. 2).

Alteraciones metabólicas: factores implicados
El metabolismo del paciente canceroso puede experimentar importantes alteraciones como consecuencia de la presencia del tumor. A pesar de la malnutrición que acompaña al crecimiento tumoral avanzado, estos cambios son en muchos aspectos diferentes a los que se presentan típicamente en las situaciones de ayuno, siendo más parecidos a los que tienen lugar en respuesta a una situación de inflamación, infección o lesiones traumáticas. De este modo, una de las principales características de la caquexia cancerosa es el desgaste tisular que sufre el paciente, que afecta particularmente al músculo esquelético y al tejido adiposo, mientras que otros órganos (hígado, bazo, riñón y adrenales) pueden llegar incluso a aumentar su peso transitoriamente. Las alteraciones metabólicas representan el aspecto más peculiar e importante de la caquexia cancerosa, ya que incluso en ausencia de malnutrición pueden determinar por sí mismas un balance energético y nitrogenado negativo, junto con un grave deterioro del organismo. Por otra parte, si bien la anorexia es un componente casi universal del síndrome caquéctico, ella sola no explica el notable desgaste observado en el paciente, ya que los patrones de pérdida de peso y los cambios en la composición corporal son diferentes de los característicos de la inanición, y sus efectos catabólicos tampoco pueden revertirse mediante la administración de un complemento calórico. De este modo, prevalece el punto de vista de que la caquexia cancerosa es principalmente debida a las alteraciones metabólicas producidas por la presencia del tumor. Entre éstas, se observa en el paciente tumoral una incrementada lipólisis, favorecida por una disminución de la actividad lipoproteína lipasa (LPL) del tejido adiposo blanco (TAB) y que propicia un aumento de los niveles de triacilgliceroles (TAG) circulantes; asimismo, se produce un aumento en el recambio proteico tisular y una incrementada utilización hepática del lactato, generado en grandes cantidades por el tumor. Durante mucho tiempo se pensó que estos desajustes metabólicos eran causados por algún factor segregado por el propio tumor, o bien como resultado de la competencia por los nutrientes entre las células del tumor y las del paciente, pero en la actualidad se sabe que estos factores proceden principalmente de la respuesta del paciente al crecimiento tumoral1, 2.
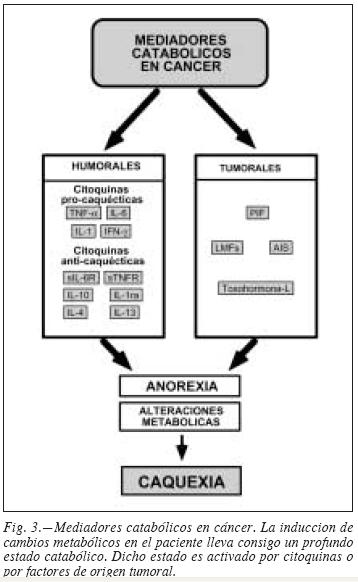
Se han dedicado notables esfuerzos encaminados a identificar el factor o factores responsables de la caquexia, debido al indudable interés clínico del tema. De los posibles mediadores identificados hasta la fecha, podemos establecer una clasificación atendiendo a su origen, pudiendo diferenciar entre mediadores de origen humoral (principalmente citoquinas) y de origen tumoral (fig. 3).
Factores humorales
Factor necrótico tumoral- α(TNF- α)
El TNF-α fue identificado en 1985 como una proteína sérica presente en animales tratados con endotoxina y que era capaz de inducir necrosis hemorrágica en tumores experimentales, viéndose poco después que era también la molécula responsable del síndrome de la caquexia asociada a la infección crónica, por lo que también fue bautizada como caquectina3. El TNF-α sintetizado principalmente por los macrófagos en respuesta a distintos estímulos invasivos, como una proteína de 26 kDa ligada a la membrana, que por proteólisis da lugar a una forma madura de 17 kDa, si bien su forma biológicamente activa es el trímero (51 kDa). Dicha molécula puede ser reconocida por dos tipos de receptores: de tipo 1 (TNFR1 o p55) y de tipo 2 (TNFR2 o p75). El TNF- αes un factor pleiotrópico, con variados efectos sobre el crecimiento celular, angiogénesis, citotoxicidad, inflamación e inmunomodulación.
Si el TNF-α fuese el mediador responsable de la caquexia cancerosa, esta citoquina debería ser capaz de mimetizar los diferentes aspectos que caracterizan dicho síndrome, tanto a nivel de pérdida de peso corporal como en los distintos cambios metabólicos observados. En este sentido, la administración episódica de TNF-α no es capaz de inducir caquexia (debido a la aparición de taquifilaxis), por lo que su efecto es mayor cuando se administra en dosis escaladas. En esta misma línea cabe destacar los experimentos realizados por el grupo de A. Oliff por medio de la implantación de células CHO que expresaban constitutivamente el gen del TNF-α humano en ratones inmunodeprimidos nude, observando una masiva pérdida de peso en los mismos asimilable a un proceso caquéctico4.
Por otra parte, los niveles circulantes de TNF-α están aumentados en más de la mitad de los pacientes con infecciones parasíticas (malaria y leishmaniasis) y con septicemia, situaciones que están asociadas a una fuerte caquexia, y que en este último caso sería atribuible a los elevados niveles de endotoxina (lipopolisacárido o LPS), componente bacteriano cuya administración a individuos sanos produce un aumento en los niveles plasmáticos de TNF-α. Las concentraciones circulantes de TNF-α en pacientes cancerosos son más variables, con altos niveles en un 50% de los pacientes estudiados, así como también en niños con leucemia linfoblástica aguda; en cambio, otros estudios no han hallado tal aumento. En los modelos experimentales la situación es similar, es decir, proporcionan resultados controvertidos. La explicación a estos resultados contradictorios puede atribuirse a las diferencias entre las distintas metodologías utilizadas, a la corta vida media del TNF-α in vivo, o a la producción paracrina de la citoquina.
Por otra parte, los efectos metabólicos inducidos por la administración de TNF-α son similares a los observados en situaciones de caquexia5. El TNF-α disminuye los niveles de mRNA y la actividad de la LPL en el TAB, tanto in vitro (salvo en adipocitos humanos) como in vivo en diferentes especies, lo que se relaciona con una menor captación de 14C-trioleína y unos mayores niveles de lípidos circulantes (principalmente TAG). También se ha visto que el TNF-α aumenta la lipólisis en células 3T3 en cultivo. La hipertrigliceridemia inducida por TNF-α puede ser debida también a una mayor lipogénesis hepática y la correspondiente producción de lipoproteínas de muy baja densidad (VLDL). El TNF-α también activa la lipólisis en el TAB e inhibe el transporte de glucosa en los adipocitos y por lo tanto su disponibilidad como substrato lipogénico), aunque no afecta la lipogénesis en el TAB, donde tan sólo se ha observado una disminución en la actividad de la acetil-CoA carboxilasa, enzima clave en el control de la lipogénesis6. Por otra parte, se ha observado que el tratamiento con anticuerpos anti-TNF-α es capaz de revertir parcialmente estas anomalías metabólicas7.
Existen asimismo numerosas evidencias experimentales a favor de un papel del TNF-α en la pérdida de proteína muscular. El tratamiento crónico con TNF-α provoca una disminución de la proteína corporal, especialmente en el músculo esquelético, y también aumenta la tasa de degradación proteica tanto in vivo como in vitro. En nuestro laboratorio hemos podido observar que el tratamiento crónico con TNF-α aumenta la tasa de degradación proteica en el músculo, fenómeno asociado a una activación del sistema proteolítico dependiente de ATP y ubiquitina8. Este sistema proteolítico citosólico implica la conjugación covalente y dependiente de energía del péptido ubiquitina con aquellas proteínas que han de ser degradadas, que se convierten así en sustrato reconocido por las proteasas del complejo proteasoma 26S. Este mecanismo permite la degradación tanto de proteínas estructurales como funcionales, y se encuentra activado en el musculo esquelético de animales en diferentes situaciones patológicas, como la caquexia cancerosa, las infecciones, la acidosis o la atrofia. También hemos visto que la administración de TNF-α aumenta el grado de ubiquitinización de las proteínas musculares, lo que constituye la etapa previa a su degradación por el mencionado sistema, así como un aumento en la expresión de diferentes genes asociados a este sistema. Por otra parte, el tratamiento con anticuerpos anti-TNF-α es capaz de revertir en gran medida los cambios atribuibles al estado caquéctico. Asimismo, hemos comprobado que esta acción es directa, lo que puede explicarse por la presencia de los dos tipos de receptores del TNF-α en el músculo esquelético. Más recientemente hemos podido verificar que en situaciones de caquexia disminuye no sólo el contenido proteico muscular, sino también el de DNA, hecho explicable por la existencia de un proceso de fragmentación del DNA relacionado con fenómenos de apoptosis9, y que el TNF-α también puede mimetizar esta respuesta10.
El TNF-α se ha asociado también a situaciones de resistencia a la insulina. En este sentido, las situaciones patológicas asociadas a una alta producción de TNF-α (endotoxemia, cáncer, trauma) muestran asimismo resistencia periférica a la insulina, y la administración de TNF-α a individuos sanos es capaz de reducir la sensibilidad a la insulina. El TNF-α ha sido implicado en la diabetes mellitus de tipo II, y también se ha visto una producción aumentada de esta citoquina por parte del TAB en diferentes modelos de obesidad animal, así como una sobreexpresión en músculo esquelético de individuos diabéticos. Finalmente, cabe constatar que la administración de TNF-α (tanto periférica como intracerebral) incrementa el flujo sanguíneo y la actividad termogénica del tejido adiposo marrón (TAM), asociada con la proteína desacopladora UCP1. Durante el estado caquéctico, existe un incremento en la termogénesis del TAM observada tanto en humanos como en animales experimentales. La UCP1, presente exclusivamente en el TAM, es una proteína transportadora que genera calor mediante la disipación del gradiente de protones creado a través de la membrana mitocondrial interna durante la respiración, desacoplando de esta manera la respiración de la síntesis de ATP. Más recientemente se han descrito otras proteínas de la misma familia, como la UCP2 y la UCP3. La UCP2 se expresa ubicuamente, mientras que la UCP3 se expresa abundantemente en músculo esquelético en humanos y roedores, y en el TAM en roedores. Nuestro grupo ha demostrado que durante el crecimiento tumoral existe un incremento en la expresión de los mRNAs de UCP2 y UCP3 en el músculo esquelético11, y que el TNF-α mimetiza este incremento en la expresión génica12. Además, el TNF-α es capaz de inducir el desacoplamiento de la respiración en mitocondrias aisladas13.
Otras citoquinas
Aunque las distintas evidencias acumuladas hasta la fecha apuntan a que el TNF-α podría ser el responsable de la mayor parte de los efectos metabólicos observados en situaciones de caquexia, ello no descarta que otros mediadores humorales pudieran también estar implicados. En este sentido, otras citoquinas parecen también tener un papel en este complejo síndrome14.
La interleuquina-6 (IL-6) podría ser una de estas citoquinas, por cuanto la administración de anticuerpos anti-IL-6 a ratones portadores de un adenocarcinoma de colon revierte los parámetros asociados al estado caquéctico; sin embargo, otros estudios similares no han podido corroborar estos resultados. Por otra parte, la IL-6 tampoco tiene un efecto sobre la proteólisis muscular in vitro, si bien sí que es capaz de estimular la gluconeogénesis en hepatocitos.
Otra citoquina que podría tener un papel en la caquexia es el interferón-γ (IFN-γ), por cuanto muestra efectos similares (y sinérgicos) a los del TNF-α. Así, se ha visto que puede inhibir la actividad LPL en células 3T3L1 y la lipogénesis en adipocitos, y que la administración de anticuerpos monoclonales anti-IFN-γ es capaz de revertir notablemente el estado caquéctico en ratones portadores del carcinoma pulmonar de Lewis. Además, la implantación en ratones nude de células CHO que producen IFN-γ de manera constitutiva provoca un fuerte estado caquéctico.
También se ha sugerido un papel significativo para el factor inhibidor de la leucemia (LIF), ya que se ha visto que tumores que secretan LIF en ratón pueden provocar una respuesta caquéctica. Otros candidatos a considerar serían el factor de crecimiento transformante-β (TGF-β), el factor neurotrófico ciliar (CNTF)(producido por la glía, pero también por músculo esquelético) y la interleuquina-1 (IL-1), la cual tiene fuertes efectos anorexigénicos y pirogénicos, aunque se ha visto que la administración del antagonista del receptor de la IL-1 (IL-1ra) a ratas caquécticas no revierte la caquexia.
En resumen, el TNF-α parece tener un papel importante en la caquexia, pero no debe descartarse que otras citoquinas puedan tener también un papel significativo, sin olvidar los cambios en el estado hormonal (fig. 4).

Factores tumorales
Diferentes estudios han puesto de manifiesto que, al margen de las citoquinas, otras moléculas mediadoras de origen tumoral también podrían tener un papel (fig. 3). La primera en ser descrita fue la toxohormona L, al observarse que extractos de células del carcinoma Krebs-2 de ratón podían inducir caquexia cuando se inyectaban a animales sanos; posteriormente se aisló esta sustancia a partir de líquido ascítico de pacientes con hepatoma y de ratones portadores de sarcoma, observándose que es un polipéptido de 75 kDa capaz de inducir movilización lipídica e inmunosupresión.
El grupo de M. Tisdale aisló un compuesto a partir de ratones portadores del adenocarcinoma de colon MAC16, y más tarde lo obtuvieron a partir de la orina de pacientes cancerosos15. Dicho compuesto fue denominado factor movilizador de lípidos (LMF), dado que era capaz de inducir lipólisis en TAB a través de la activación de la adenilato ciclasa, y también de activar la glucogenólisis hepática por la misma vía.
El factor inductor de anemia (AIS) es una proteína de 50 kDa segregada por tumores malignos que deprime las funciones de la células inmunocompetentes, y también es capaz de reducir la ingesta, el peso corporal y la grasa corporal en conejos, así como de presentar una importante actividad lipolítica.
Un último compuesto incorporado a esta relación es el factor inductor de proteólisis (PIF), un proteoglicano de 24 kDa producido por un adenocarcinoma de colon murino16 y que también está presente en pacientes cancerosos (pero no así en individuos sanos o en pacientes cancerosos con poca pérdida de peso), y que podría estar implicado en la incrementada degradación proteica muscular y en la disminuida síntesis proteica, activando la degradación proteica a través del sistema dependiente de ATP y ubiquitina, aunque por el contrario no produce anorexia. La administración de este compuesto a animales sanos provoca desgaste muscular. Su acción puede ser inhibida in vivo por el ácido eicosapentaenoico (EPA), lo cual indica que podría actuar a través de metabolitos del araquidónico como el 15-HETE. También se ha visto que estimula la síntesis de proteínas de fase aguda a través de la vía de los factores de transcripción NF-κB y STATS3.
El hecho que algunos de estos factores (caso del LMF y el PIF) mimeticen los efectos de la caquexia sobre el metabolismo lipídico y proteico, y también el que estén presentes en la orina de pacientes cancerosos, hace pensar en un posible papel en la caquexia en humanos.
Por lo tanto, hasta la fecha no se ha podido identificar el mediador responsable de la caquexia, aunque a la vista de los resultados el TNF-α podría ser un buen candidato, pues mimetiza las alteraciones metabólicas (pérdida peso, anorexia, termogénesis, resistencia a la insulina, desgaste proteico muscular y movilización lipídica en el TAB), pero no puede explicar todos los casos, por lo que posiblemente la activación de la caquexia sea una combinación de diferentes factores.
Referencias
1. Evans RD, Argilés JM, Williamson DH: Metabolic effects of tumour necrosis factor-alpha (cachectin) and interleukin-1. Clin Sci 1989; 77:357-64. [ Links ]
2. Argilés JM, García-Martínez C, Llovera M, López-Soriano FJ: The role of cytokines in muscle wasting: its relation with cancer cachexia. Med Res Rev 1992; 12:637-52. [ Links ]
3. Beutler BA, Milsark IW, Cerami A: Cachectin/tumor necrosis factor: production, distribution, and metabolic fate in vivo. J Immunol 1985; 135:3972-7. [ Links ]
4. Oliff A, Defeo-Jones D, Boyer M, Martínez D, Kiefer D, Vuocolo G y cols.: Tumors secreting human TNF/cachectin induce cachexia in mice. Cell 1987; 50:555-63. [ Links ]
5. García-Martínez C, Costelli P, López-Soriano FJ, Argilés JM: Is TNF really involved in cachexia? Cancer Invest 1997; 15:47-54. [ Links ]
6. Argilés JM, López-Soriano J, Busquets S, López-Soriano FJ: Journey from cachexia to obesity by TNF. FASEB J 1997; 11:743-51. [ Links ]
7. Costelli P, Carbó N, Tessitore L, Bagby GJ, López-Soriano FJ, Argilés JM y cols.: Tumor necrosis factor-alpha mediates changes in tissue protein turnover in a rat cancer cachexia model. J Clin Invest 1993; 92:2783-9. [ Links ]
8. Argilés JM, López-Soriano FJ: The ubiquitin-dependent proteolytic pathway in skeletal muscle: its role in pathological states. Trends Pharmacol Sci 1996; 17:223-6. [ Links ]
9. Van Royen M, Carbó N, Busquets S, Alvarez B, Quinn LS, López-Soriano FJ y cols.: DNA fragmentation occurs in skeletal muscle during tumor growth: a link with cancer cachexia. Biochem. Biophys Res Commun 2000; 270:533-7. [ Links ]
10. Carbó N, Busquets S, van Royen M, Alvarez B, López-Soriano FJ, Argilés JM: TNF-alpha is involved in activating DNA fragmentation in skeletal muscle. Br J Cancer 2002; 86:1012-6. [ Links ]
11. Sanchís D, Busquets S, Álvarez B, Ricquier D, López-Soriano FJ, Argilés JM: Skeletal muscle UCP2 and UCP3 gene expression in a rat cancer cachexia model. FEBS Lett 1998; 436:415-8. [ Links ]
12. Busquets S, Sanchís D, Álvarez B, Ricquier D, López-Soriano FJ, Argilés JM: In the rat, tumor necrosis factor alpha administration results in an increase in both UCP2 and UCP3 mRNAs in skeletal muscle: a possible mechanism for cytokine-induced thermogenesis? FEBS Lett 1998; 440:348-50. [ Links ]
13. Busquets S, Aranda X, Ribas-Carbó M, Azcón-Bieto J, López- Soriano FJ, Argilés JM: Tumour necrosis factor-alpha uncouples respiration in isolated rat mitochondria. Cytokine 2003; 22:1-4. [ Links ]
14. Argilés JM, Álvarez B, López-Soriano FJ: The metabolic basis of cancer cachexia. Med Res Rev 1997; 17:477-98. [ Links ]
15. Hirai K, Ishiko O, Tisdale M: Mechanism of depletion of liver glycogen in cancer cachexia. Biochem Biophys Res Commun 1997; 241:49-52. [ Links ]
16. Todorov PT, Field WN, Tisdale MJ: Role of a proteolysis-inducing factor (PIF) in cachexia induced by a human melanoma (G361). Br J Cancer 1999; 80:1734-7. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Josep. M. Argilés
Departamento de Bioquímica y Biología Molecular
Facultad de Biología
Universidad de Barcelona
Diagonal, 645.
08028 Barcelona
E-mail: argiles@porthos.bio.ub.e

