










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.18 no.4 abr. 2001
REVISIÓN DE CONJUNTO
La homocisteína. ¿El factor de riesgo cardiovascular del próximo milenio?
I. Suárez García, J.F. Gómez Cerezo, J.J. Ríos Blanco, F.J. Barbado Hernández, J.J. Vázquez Rodríguez
Servicio de Medicina Interna. Hospital Universitario La Paz. Madrid.
RESUMEN
Varios estudios epidemiológicos han demostrado que la hiperhomocisteinemia es un factor de riesgo de arteriosclerosis coronaria, cerebral, periférica y aórtica. Este riesgo es independiente de otros factores de riesgo cardiovascular, y es mayor cuanto mayor sea la concentración de homocisteína. Sin embargo, los estudios prospectivos muestran resultados aún contradictorios. La hiperhomocisteinemia se asocia también a un mayor riesgo de enfermedad tromboembólica que podría estar modulado por otras alteraciones de la coagulación como el factor V Leiden.
El aumento de la concentración de homocisteína plasmática puede deberse tanto a alteraciones hereditarias de los enzimas que intervienen en su metabolismo como a otros factores (principalmente dietéticos, como déficit de vitaminas B6, B12 y ácido fólico). El aporte de suplementos de vitamina B6, B12 y ácido fólico se ha demostrado eficaz para disminuir la concentración de homocisteína plasmática. Se precisan aún ensayos clínicos que demuestren que la disminución de homocisteína plasmática conlleva una disminución del riesgo de enfermedad cardiovascular.
PALABRAS CLAVE: Arteriosclerosis. Hiperhomocisteinemia. Homocisteína. Homocistinuria. Trombosis venosa. Enfermedad cardiovascular.
Homocysteine. The cardiovascular risk factor of the next millenium?
ABSTRACT
Several epidemiologic studies have demonstrated that hyperhomocysteinemia is a risk factor for arteriosclerosis in coronary, cerebral, peripheral and aortic arteries. This risk is independent of other cardiovascular risk factors, and it is dose related. However, prospective studies show contradictory findings. Hyperhomocysteinemia is also associated with a higher risk of venous thrombosis to which other coagulation disorders, such as factor V Leiden, could contribute.
Hyperhomocysteinemia can be due to genetic defects in the enzymes that control homocysteine metabolism, and also to other factors, mainly nutritional (deficiencies in vitamin B6, vitamin B12, or folic acid). Dietary supplements of these vitamins reduce plasma homocysteine levels. Randomized clinical trials are still needed to demonstrate that reducing plasma homocysteine levels will reduce the risk for cardiovascular disease.
KEY WORDS: Arteriosclerosis. Homocysteine. Hyperhomocysteinemia. Homocystinuria. Venous thrombosis. Cardiovascular disease.
Trabajo aceptado: 2 de Junio de 2000
Correspondencia: I. Suárez García. Servicio de Medicina Interna. Hospital Universitario La Paz. Paseo de la Castellana, 261. 28046 Madrid.
UNA INTRODUCCIÓN AL METABOLISMO DE LA HOMOCISTEÍNA
La homocisteína es un aminoácido que se sintetiza en el organismo a partir de otro: la metionina. La única fuente de metionina es la ingesta, a partir principalmente de proteínas animales.
El metabolismo de la homocisteína está muy relacionado con las vitaminas B6, B12 y ácido fólico. La metionina que proviene de la ingesta se metaboliza, principalmente en el hígado, en homocisteína (Figura 1). A partir de aquí, la homocisteína puede seguir dos vías:

a) Remetilación: transformarse de nuevo en metionina, mediante un proceso catalizado por la enzima metionina sintasa y dependiente de la vitamina B12 y del N5-metil-tetrahidrofolato (que actúan como cofactor y como dador de metilo).
b) Transulfuración: unirse a la serina y transformarse en cisteína, que se elimina por la orina. Esta segunda vía depende de la enzima cistationina β-sintasa, que tiene a la vitamina B6 como cofactor (88).
LA HOMOCISTINURIA
Las extremidades longilíneas, aracnodactilia, genu valgo, pie plano e hiperlaxitud articular conforman un fenotipo denominado marfanoide. Éste no es exclusivo de la enfermedad de Marfan, sino que se observa también en otra enfermedad hereditaria: la homocistinuria.
La homocistinuria se produce por diversos defectos enzimáticos en la vía metabólica de la homocisteína, que se heredan de forma autosómica recesiva. La consecuencia es un acúmulo de homocisteína, que es la responsable de las manifestaciones clínicas de esta enfermedad (12).
El exceso de homocisteína interfiere en el entrecruzamiento normal del colágeno. El depósito de colágeno alterado en el ligamento suspensorio del cristalino y la matriz ósea explica que el 90% presenten luxación del cristalino y la osteoporosis sea frecuente (12).
La alteración enzimática más frecuentemente implicada es la deficiencia de cistationina β-sintasa, con una prevalencia de 1 caso por 200.000 habitantes (88). La consecuencia de este trastorno es un acúmulo de homocisteína y metionina. Es de destacar, además del fenotipo característico, que la principal causa de mortalidad son las complicaciones aterotrombóticas, que ocurren ya desde la primera década de la vida. El 25% de los homocigotos fallecen antes de los 30 años debido a complicaciones cardiovasculares: trombosis arteriales (cerebral, periférica y coronaria) y enfermedad tromboembólica venosa (47,53). La mitad de los pacientes presenta retraso mental y gran parte tienen alteraciones de la conducta. Este hecho se ha intentado explicar bien por accidentes cerebrovasculares recurrentes o bien por un efecto tóxico directo de la homocisteína sobre las células cerebrales (12).
El tratamiento del déficit de cistationina β-sintasa debe iniciarse precozmente con la restricción de metionina de la dieta y la administración de suplementos de cistina. En el 50% de los pacientes la actividad de esta enzima es indetectable, y en el resto conserva parcialmente su actividad. Este último grupo responde a suplementos orales de piridoxina produciéndose un descenso de la concentración plasmática de homocisteína. Se ha observado que este 50% que responde a la piridoxina tiene menor número de complicaciones cardiovasculares (12,53).
Otra enfermedad infrecuente es la deficiencia de la enzima metilentetrahidrofolato reductasa de la que se han descrito sólo 25 casos. Esta enzima interviene en la formación de metionina a partir de la homocisteína y en la formación de tetrahidrofolato, que a su vez es necesario para la síntesis de ADN y ARN. Como consecuencia se producen también alteraciones en el sistema nervioso: desde alteraciones de la conciencia hasta retraso mental profundo. Esta deficiencia tiene peor pronóstico ya que no se ha demostrado una terapia eficaz, aunque se ha observado en algunos casos respuesta a folato oral (88).
Existen otras deficiencias menos frecuentes como la de metionina sintasa y defectos en el metabolismo de la vitamina B12 (88).
¿QUÉ OCURRE CON LOS HETEROCIGOTOS?
Hasta ahora hemos visto que los pacientes con homocistinuria -los homocigotos para todos estos defectos enzimáticos- tienen un riesgo elevado de complicaciones vasculares. Actualmente se admite también en los heterocigotos un mayor riesgo de enfermedad oclusiva vascular.
Los heterocigotos, generalmente asintomáticos, representan una parte importante de la población general. Los heterocigotos para la deficiencia de cistationina β-sintasa representan entre el 1 y el 2% de la población. Estos sujetos presentan concentraciones de homocisteína plasmática discretamente elevadas, o bien concentraciones normales que se elevan de forma exagerada tras una sobrecarga de metionina oral.
Existe una variante termolábil de la enzima metilentetrahidrofolato reductasa que produce concentraciones elevadas de homocisteína en sangre. Esta variante es ocasionada por una mutación especialmente frecuente en la población francófona de Canadá (38%), y en el 5 al 15% del total de la población canadiense. Los homocigotos para esta mutación tienen un aumento exagerado de la homocisteinemia en respuesta a la depleción de ácido fólico, y aunque no tienen mayor riesgo de complicaciones cardiovasculares sí podrían tenerlo si se asocia un déficit de ácido fólico (4,11,21).
DIAGNÓSTICO
El hallazgo de homocisteína en la orina no es específico de esta enfermedad ya que puede encontrarse también en otras enfermedades, e incluso en sujetos normales como un artefacto por contaminación bacteriana (12).
El diagnóstico se obtiene determinando la homocisteína plasmática total en ayunas, cuyo valor normal oscila entre 5 y 15 micromoles/litro (18). En los heterocigotos la concentración de homocisteína plasmática puede ser normal; en estos casos se hace una prueba de sobrecarga oral de metionina que provoca un aumento de la concentración plasmática de homocisteína. Esta prueba ha sido criticada ya que es poco útil en las alteraciones de la metilentetrahidrofolato reductasa (88).
OTRAS CAUSAS DE HIPERHOMOCISTEINEMIA
Además de estas alteraciones hereditarias, existen otras causas de aumento de la concentración de homocisteína.
Los más importantes son los factores dietéticos. La homocisteína se produce a partir de la metionina, y la principal fuente de metionina son las proteínas de origen animal: carne, huevos y leche. Las verduras, cereales o legumbres aportan menor cantidad de proteínas y además las proteínas vegetales tienen una menor proporción de metionina.
Recordemos además que las vitaminas B6, B12 y ácido fólico son necesarias como cofactores para el metabolismo de la homocisteína, bien para transformarla de nuevo en metionina o bien para transformarla en cisteína y otros compuestos que se excretan por la orina. Estas vitaminas son muy sensibles a la destrucción por medios físicos o químicos que se emplean en el procesado y conservación de los alimentos. Por tanto una dieta con una inadecuada ingestión de estas vitaminas, rica en alimentos de origen animal o altamente refinados condicionará un aumento de la homocisteína en sangre (71,81).
Otras causas de aumento de la homocisteína se exponen en la tabla I.

PAPEL DE LA HOMOCISTEíNA EN LA ARTERIOSCLEROSIS Y LA ENFERMEDAD TROMBOEMBÓLICA
Una vez conocidos todos estos datos, llega el momento de plantearnos algunas cuestiones:
--¿Cuál es el mecanismo responsable de las complicaciones aterotrombóticas en los pacientes con homocistinuria?
--¿Cabría esperar un aumento de morbimortalidad cardiovascular en pacientes con aumento moderado de la homocisteína?
Es decir, estamos preguntándonos si la homocisteína juega un papel en la arteriosclerosis y en la enfermedad tromboembólica.
ESTUDIOS EN ANIMALES
En 1969, McCully inyectó homocisteína tiolactona subcutánea en un grupo de conejos y observó que a las tres semanas se habían formado placas de aterosclerosis en las arterias coronarias (48). Posteriormente repitió el experimento con dosis mayores durante un mes y observó que se formaban también trombos en las venas de las patas, del abdomen y de los pulmones. Estos trombos no se producían en los conejos que habían recibido simultáneamente inyecciones de vitamina B6 (49).
Experimentos posteriores realizados en otros animales y con homocisteína aportada con la dieta demostraron que un aumento en la concentración de homocisteína plasmática mediante ingestión, inyección, o asociando dietas carenciales en vitamina B6 reproducían las características de la arteriosclerosis que se observaba en niños con homocistinuria hereditaria.
Se ha observado que las células cultivadas de niños con homocistinuria muestran un patrón de crecimiento anormal, semejante en algunos aspectos al de las células cancerosas. Estos datos sugieren que la homocisteína estimula el crecimiento de las células, lo cual también concuerda con el hecho de que los niños con homocistinuria crezcan rápidamente en la infancia, y tengan las extremidades más alargadas, alcanzando a veces mayor estatura que sus familiares (46,49).
ESTUDIOS EN HUMANOS
Desde estos primeros experimentos en animales, se han venido haciendo numerosos estudios en humanos. Más de 20 estudios de casos y controles y transversales han demostrado que existe una asociación clara entre la elevación de la homocisteína plasmática y el riesgo de enfermedad vascular periférica, cerebral y coronaria.
Entre ellos, Selhub et al realizaron un estudio transversal con más de 1000 sujetos del estudio Framingham demostrando la correlación de la concentración de homocisteína con el grado de estenosis de la arteria carótida, incluso después de ajustar otros factores de riesgo cardiovascular (70). Se ha demostrado también una correlación positiva con el grado de arteriosclerosis coronaria (52,85), periférica (5) y aórtica (38), y la asociación con el riesgo de enfermedad coronaria de inicio precoz (58).
Sin embargo, aunque los estudios epidemiológicos han mostrado una fuerte asociación entre la hiperhomocisteinemia y la enfermedad cardiovascular, los datos de los estudios prospectivos realizados hasta ahora son todavía contradictorios (23).
Entre los primeros estudios prospectivos a gran escala que relacionaban la homocisteína con la enfermedad coronaria destaca el Physicians´ Health Study de 1992 (Stampfer et al), realizado en una población de más de 14.000 médicos varones; a los sujetos de estudio se les determinó la homocisteinemia y se les siguió durante 5 años. Los que tenían una concentración superior al 12% sobre el límite de la normalidad tuvieron un riesgo 3 veces mayor de infarto de miocardio, incluso después de haber corregido otros factores de riesgo. El estudio concluía que el 7% de los 271 infartos registrados se podía atribuir a la hiperhomocisteinemia (72). Tres años después, Arneson et al obtuvieron resultados similares con otro estudio realizado en Noruega con más de 20.000 pacientes (3).
Además de estos dos estudios, en el metanálisis realizado por Eikelboom et al (16) se citan otros seis estudios prospectivos que concluían que la hiperhomocisteinemia es un factor de riesgo independiente de enfermedad vascular (8,56,60,61,75,86). Sin embargo, se citan también otros seis estudios prospectivos que no han podido confirmar estos resultados. Tres de ellos fueron realizados en la misma población de médicos varones del Physicians´ Health Study ya citado, pero tras un periodo de seguimiento más prolongado, y no consiguieron demostrar una asociación entre la homocisteinemia y el riesgo de enfermedad coronaria o infarto (1,14,17,20,83,84).
Entre los últimos estudios de cohortes realizados, Bostom et al estudiaron sujetos ancianos del estudio Framingham, hallando que la concentración de homocisteína era un factor de riesgo independiente de accidente cerebrovascular, mortalidad cardiovascular y mortalidad global (6,7).
Para resumir todos estos datos, los estudios epidemiológicos han demostrado que la hiperhomocisteinemia es un factor de riesgo de arteriosclerosis independiente de otros factores como tabaco o colesterol, y además tiene un efecto multiplicativo con otros factores como tabaco e hipertensión arterial (24). Cifras mayores de 14 micromoles/litro representan mayor riesgo de arteriosclerosis, y el riesgo es mayor cuanto mayor sea la concentración de homocisteína. Se ha estimado que el 10% del riesgo de enfermedad coronaria en la población general es atribuible a la homocisteína (9). Por ahora se acepta esta asociación con ciertas reservas, principalmente por dos razones: en primer lugar, los estudios prospectivos muestran resultados contradictorios, y en segundo lugar porque no se han realizado aún ensayos clínicos que demuestren que la disminución de la homocisteína plasmática conlleve una disminución del riesgo de enfermedad cardiovascular.
¿CUÁL ES EL MECANISMO POR EL QUE LA HOMOCISTEÍNA PRODUCIRÍA ARTERIOSCLEROSIS?
De nuevo aquí los primeros estudios se hicieron en animales. En 1976 Harker et al administraron homocisteina intravenosa a babuinos. El estudio anatomopatológico mostró descamación del endotelio de la aorta, engrosamiento de la íntima, acúmulo de lípidos y proliferación de células musculares lisas, así como disminución de la vida media plaquetaria, y ya entonces se propuso que la homocisteína podía tener un efecto tóxico directo sobre el endotelio (27).
La homocisteína en el plasma se oxida (formando homocistina, homocisteína tiolactona y otros compuestos) y en este proceso se forman peróxido de hidrógeno y radicales libres como el hidroxilo y el anión superóxido, que son muy reactivos y producen lesión endotelial (25,51,68,73,87,88,89) (Figura 2). El endotelio dañado a su vez hace que quede expuesta la matriz subendotelial, con lo que se estimula la agregación plaquetaria (27,28).
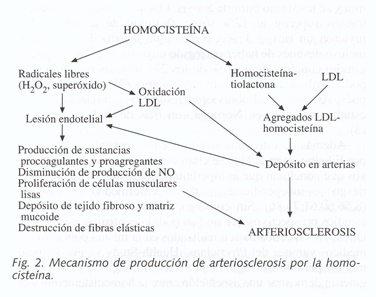
Los radicales libres son capaces de producir una oxidación de los lípidos, y entre ellos las lipoproteínas de baja densidad (LDL). Estas LDL oxidadas tienen un efecto tóxico sobre la pared vascular y son más fácilmente reconocidas y captadas por los macrófagos del endotelio (29,32,33,40,59,68).
Por otra parte, cuando la concentración de homocisteína está aumentada, se favorece la formación de una forma muy reactiva, la homocisteína tiolactona, que produce en el hígado agregados con las LDL. Estos agregados presentan una mayor afinidad por la fibrina que las LDL y van a la sangre donde son captados por los macrófagos de la pared arterial que los degradan, liberando lípidos y colesterol que se depositan en las placas de ateroma. La homocisteína tiolactona también altera el metabolismo de las células endoteliales, favoreciendo la formación de más radicales libres y produciendo más daño endotelial (45,54).
Otras alteraciones que se producen son: disminución de la producción de óxido nítrico (40,89); proliferación de las células musculares lisas de la pared arterial (76,77); alteración de la síntesis del colágeno y depósito de matriz glicoproteica extracelular en la pared de los vasos (13,45,46); y destrucción de fibras elásticas de la pared vascular (13,45,46).
PAPEL DE LA HOMOCISTEÍNA EN LA ENFERMEDAD TROMBOEMBÓLICA
La superficie endotelial dañada expresa sustancias procoagulantes y se han descrito numerosas anomalías en el proceso de la coagulación y la agregación plaquetaria que contribuyen a una acción protrombótica de la homocisteína; entre ellas, aumento de la síntesis de tromboxano A2 (50), activación de los factores V y XII (62,66), aumento de la actividad del factor tisular (22), disminución de la actividad de la proteína C (65) y del activador tisular del plasminógeno (26), inhibición de la expresión de trombomodulina en la superficie celular (39), así como aumento de la activación plaquetaria (27,28).
Considerando esta acción protrombótica, se ha intentado también estudiar el papel de la homocisteína en la trombosis venosa. En 1996 se publicó un estudio de casos y controles en que se demostraba que la hiperhomocisteinemia moderada es un factor de riesgo para la trombosis venosa en la población general (30).
Desde entonces hay controversia: existen estudios que lo confirman y otros que no han encontrado ninguna asociación. Por otra parte, sólo un tercio de los pacientes con homocistinuria tienen trombosis (42). ¿Por qué existe esta variabilidad? Las diferencias podrían deberse a que la homocisteína elevada podría ser tan sólo un factor de riesgo débil, a menos que exista un defecto de la anticoagulación endógeno (64). En esta línea, lo que más se ha estudiado es el papel que podría jugar la resistencia a la proteína C activada, ya que es la causa más frecuente de trombofilia familiar.
La proteína C activada es un anticoagulante fisiológico que se produce en la superficie de las células endoteliales, y que inactiva a los factores VIIIa y Va. La mayoría de los casos de resistencia a la proteína C activada son producidos por una mutación en el gen del factor V que produce la sustitución de un aminoácido en el factor Va precisamente en uno de los lugares en que se une a la proteína C. Esta variedad se denomina factor V Leiden, y se encuentra en el 30-60% de los casos de trombofilia familiar y en el 3-7% de la población sana. El riesgo de trombosis se multiplica por 80 en homocigotos y por 7 en heterocigotos. La mayor parte de las trombosis se producen cuando coexisten otros defectos genéticos (como déficit de proteína C o S, o mutaciones de la protrombina) o circunstancias precipitantes como inmovilización (42, 67, 74).
Varios autores han estudiado la relación entre la hiperhomocisteinemia, el factor V Leiden y el riesgo de enfermedad tromboembólica. Mandel et al estudiaron varias familias con homocistinuria: de once sujetos homocigotos, seis tuvieron enfermedad tromboembólica. Todos ellos fueron homocigotos o heterocigotos para el factor V Leiden. Uno fue tratado con anticoagulantes orales y no desarrolló enfermedad tromboembólica a pesar de ser heterocigoto para el factor V Leiden. Los otros cuatro homocigotos restantes no desarrollaron enfermedad tromboembólica ni tenían factor V Leiden. Los autores concluían que la homocistinuria por sí sola no era suficiente para producir enfermedad tromboembólica, pero sí era un factor de riesgo cuando se asociaba al factor V Leiden (42).
Ridker investigó 145 sujetos con tromboembolismo venoso comparados con 646 controles y concluyó que el riesgo de tromboembolismo venoso idiopático estaba aumentado tanto en los sujetos con hiperhomocisteinemia sin factor V Leiden como en los sujetos con factor V Leiden sin hiperhomocisteinemia; pero cuando se consideraban los sujetos que tenían ambos factores de riesgo, el riesgo se multiplicaba por 3,6 (64).
En conclusión, la variabilidad en el riesgo trombótico en la hiperhomocisteinemia podría deberse al factor V Leiden. El hecho de que los dos factores de riesgo tengan un efecto multiplicativo podría deberse a que la homocisteína también induce la activación del factor V en las células endoteliales e inhibe la activación de la proteína C (64).
LA TEORÍA DE LA HOMOCISTEÍNA Y LA PROFILAXIS DE LA ENFERMEDAD CARDIOVASCULAR
Teniendo en cuenta que la mayor parte de los casos de hiperhomocisteinemia se deben a déficits de vitaminas B6, B12 y ácido fólico (71,81), se ha estudiado la eficacia de los suplementos de estas vitaminas en reducir las concentraciones plasmáticas de homocisteína. Varios ensayos clínicos han confirmado que el aporte de suplementos de ácido fólico, solo o combinado con vitamina B6 y B12, o vitamina B12 sola, reducen la concentración plasmática de homocisteína en un periodo de 2 a 6 semanas. Este efecto se ha demostrado incluso en sujetos que no tienen déficit de ninguna de estas vitaminas, y en los que tienen hiperhomocisteinemia de cualquier etiología. El tratamiento con estos suplementos vitamínicos se ha demostrado eficaz, con mínimos efectos secundarios y de bajo coste (10,31,34,37,41,69).
Sin embargo, aún está por demostrar que la reducción de la homocisteína plasmática mediante estos suplementos tenga algún efecto en la enfermedad cardiovascular. Varios ensayos clínicos se están llevando a cabo actualmente para intentar responder a esta pregunta: ¿disminuiría el aporte de suplementos con vitaminas B6, B12 y ácido fólico el riesgo de enfermedad cardiovascular? Si se demuestra esta hipótesis, tendríamos un método eficaz, seguro y barato para prevenir la enfermedad cardiovascular en la población general (16).
UTILIDAD DE LA HOMOCISTEÍNA EN EL DIAGNÓSTICO DE LA ENFERMEDAD CARDIOVASCULAR
¿Sería conveniente determinar la concentración de homocisteína plasmática del mismo modo en que se determinan otros factores de riesgo cardiovasculares como el colesterol? Por el momento no se ha demostrado que exista un beneficio en su análisis rutinario, aunque sí parece razonable descartar hiperhomocisteinemia en los pacientes con enfermedad coronaria sin otros factores de riesgo cardiovascular conocidos, o en aquellos que presenten aterosclerosis prematura (19).
CONCLUSIONES
La hiperhomocisteinemia se asocia a un aumento del riesgo de arteriosclerosis cerebral, periférica y coronaria. Esta asociación cumple los criterios de consistencia, fuerza, plausibilidad biológica y relación dosis-respuesta. Para poder aceptar que la homocisteína no sólo es un factor de riesgo sino un factor causal faltan todavía ensayos clínicos que demuestren una disminución del riesgo de arteriosclerosis al reducir la homocisteína plasmática. La hiperhomocisteinemia se asocia también a un mayor riesgo de enfermedad tromboembólica modulado por el factor V Leiden.
Si la mayor parte de los casos de hiperhomocisteinemia se deben a un déficit de vitaminas B6, B12 y fólico, y se ha observado que sus suplementos disminuyen la concentración de homocisteina plasmática, es lógico deducir que el aporte dietético de estas vitaminas disminuiría el riesgo de enfermedad vascular. Sin embargo, aún no se han completado ensayos clínicos a gran escala que lo demuestren.
Bibliografía
1. Alfthan G, Pekkanen J, Jauhiainen M, et al. Relation of serum homocysteine and lipoprotein(a) concentrations to atherosclerotic disease in a prospective Finnish population based study. Atherosclerosis 1994; 106: 9-19. [ Links ]
2. Andersson A, Brattström L, Israelsson B, Isaksson A, Hamfelt A, Hulberg B. Plasma homocysteine before and after methyonine loading with regard to age, gender and menopausal status. Eur J Clin Invest 1992; 22: 79-87. [ Links ]
3. Arnesen E, Refsum H, Bonaa KH, Ueland PM, Forde OH, Nordrehaug JE. Serum total homocysteine and coronary heart disease. Int J Epidemiol 1995; 24: 704-9. [ Links ]
4. Arruda VR, von Zuben PM, Chiaparini LC, Annichino-Bizzacchi JM, Costa FF. The mutation Ala677ÆVal in the methylenete trahydrofolate reductase gene: a risk factor for arterial disease and venous thrombosis. Thromb Haemost 1997; 77: 818-21. [ Links ]
5. Van den Berg M, Stehouwer CD, Bierdrager E, Rauwerda JA. Plasma homocysteine and severity of atherosclerosis in young patients with lower-limb atherosclerotic disease. Arterioscler Thromb Vasc Biol 1996; 16: 165-71. [ Links ]
6. Bostom AG, Rosenberg IH, Silbershatz H, et al. Nonfasting plasma total homocysteine levels and stroke incidence in elderly persons: the Framingham study. Ann Intern Med 1999; 131: 352-355. [ Links ]
7. Bostom AG, Silbershatz H, Rosenberg IH, et al. Nonfasting plasma total homocysteine levels and all-cause cardiovascular disease mortality in elderly Framingham men and women. Arch Intern Med 1999; 159: 1077-1080. [ Links ]
8. Bots ML, Launer LJ, Lindemans J, et al. Homocysteine and short-term risk of myocardial infarction and stroke in th elderly: the Rotterdam study. Arch Intern Med 1999; 159: 38-44. [ Links ]
9. Boushey CJ, Beresford SA, Omenn GS, Motulsky AG. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease: probable benefits of increasing folic acid intakes. JAMA 1995; 274: 1049-57. [ Links ]
10. Brattstrom LE, Israelsson B, Jeppsson JO, Hultberg BL. Folic acid - an innocuous means to reduce plama homocysteine. Scand J Clin Lab Invest 1988; 48: 215-21. [ Links ]
11. Brattstrom L, Wilcken DE, Ohrvik J, Brudin L. Commmon methylenetetrahydrofolate reductase gene mutation leads to hyperhomocysteinemia but not to vascular disease: the result of a meta-analysis. Circulation 1998; 98: 2520-6. [ Links ]
12. Cacciari E, Salardi S. Clinical and laboratory features of homocystinuria. Haemostasis 1989; 19 (suppl 1): 10-13. [ Links ]
13. Cacoub P, Gatel A, Sbai A, Piette JC, Godeau P. Hyperhomocystéinémie, athérosclérose et thromboses artérielles et veineuses. Ann Med Interne 1996; 147: 352-360. [ Links ]
14. Chasan-Taber L, Selhub J, Rosenberg IH, et al. A prospective study of folate and vitamin B6 and risk of myocardial infarction in US physicians. J Am Coll Nutr 1996; 15: 136-46. [ Links ]
15. Chauveau P, Chadefaux B, Coude M, et al. Hyperhomocysteinemia, a risk factor for atherosclerosis in chronic uremic patients. Kidney Int Suppl 1993; 41: S72-S77. [ Links ]
16. Eikelboom JW, Lonn E, Genest J, Hankey G, Yusuf S. Homocysteine and cardiovascular disease: a critical review of the epidemiologic evidence. Ann Intern Med 1999; 131: 363-375. [ Links ]
17. Evans RW, Shaten BJ, Hempel JD, Cutler JA, Kuller LH. Homocyst(e)ine and risk of cardiovascular disease in the Multiple Risk Factor Intervention Trial. Arterioscler Thromb Vasc Biol 1997; 17: 1947-53. [ Links ]
18. Falcón CR, Carreras LO. Hiperhomocisteinemia moderada: fisiopatología de la lesión endotelial e implicación clínica. Sangre 1998; 43: 47-54. [ Links ]
19. Fernández-Miranda C, Aranda JL, Gómez González P, Díaz-Rubio P, Estenoz J, Gómez de la Cámara A. La hiperhomocisteinemia es frecuente en pacientes con enfermedad coronaria. Estudio de 202 enfermos. Med Clin (Barc) 1999; 113: 407-410. [ Links ]
20. Folsom AR, Nieto FJ, McGovern PG, et al. Prospective study of coronary heart disease incidence in relation to fasting total homocysteine, related genetic polymorphisms, and B vitamins: the Atherosclerosis Risk in Communities (ARIC) study. Circulation 1998; 98: 204-10. [ Links ]
21. Frosst P, Blom HJ, Milos R, et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahidrofolate reaductase. Nat Genet 1995; 10: 111-3. [ Links ]
22. Fryer RH, Wilson BD, Gubler DB, Fitzgerald LA, Rodgers GM. Homocysteine, a risk factor for premature vascular disease and thrombosis, induces tissue factor activity in endothelial cells. Arterioscler Thromb 1993; 13: 1327-33. [ Links ]
23. Graham I. Homocysteine in health and disease. Ann Intern Med 1999; 131: 387-8. [ Links ]
24. Graham IM, Daly LE, Refsum HM, et al. Plasma homocysteine as a risk factor for vascular disease: the European Concerted Action Project. JAMA 1997; 277: 1775-81. [ Links ]
25. de Groot PG, Willems C, Boers GH, Gonsalves MD, van Aken WG, van Mourik JA.Endothelial cell dysfunction in homocystinuria. Eur J Clin Invest 1983; 13: 405-10. [ Links ]
26. Hajjar KA. Homocysteine-induced modulation of tissue plasminogen activator binding to its endothelial cell membrane receptor. J Clin Invest 1993; 91: 2873-9. [ Links ]
27. Harker LA, Ross R, Slichter SJ, Scott CR. Homocysteine-induced arteriosclerosis: the role of endothelial injury and platelet response in its genesis. J Clin Invest 1976; 58: 731-41. [ Links ]
28. Harker LA, Slichter SJ, Scott CR, Ross R.Homocystinemia: vascular injury and arterial thrombosis. N Engl J Med 1974; 291: 537-43. [ Links ]
29. Harpel PC, Chang VT, Borth W. Homocysteine and other sulfhydryl compounds enhance the binding of lipoprotein(a) to fibrin: a potential biochemical link between thrombosis, atherogenesis, and sulfhydryl compound metabolism. Proc Natl Acad Sci USA 1992; 89; 10193-7. [ Links ]
30. den Heijer M, Koster T, Blom HJ, et al. Hyperhomocysteinemia as a risk factor for deep-vein thrombosis. N Engl J Med 1996; 334: 759-62. [ Links ]
31. den Heijer M, Brouwer IA, Bos GM, et al. Vitamin supplementation reduces blood homocysteine levels: a controlled trial in patients with venous thrombosis and healthy volunteers. Arterioscler Thromb Vasc Biol 1998; 18: 356-61. [ Links ]
32. Heinecke JW, Kawamura M, Suzuki L, Chait A. Oxidation of low density lipoprotein by thiols: superoxide-dependent and -independent mechanisms. J Lipid Res 1993; 34: 2051-61. [ Links ]
33. Heinecke JW, Rosen H, Suzuki LA, Chait A. The role of sulfur-containing amino acids in superoxide production and modification of low density lipoprotein by arterial smooth muscle cells. J Biol Chem 1987; 262: 10098-103. [ Links ]
34. Homocysteine Lowering Trialists. Lowering blood homocysteine with folic acid based supplements: meta- analysis of randomised trials. BMJ 1998; 316: 894-8. [ Links ]
35. Hussein WI, Green R, Jacobsen DW, Faiman C. Normalization of hyperhomocysteinemia with L-thyroxine in hypothyroidsm. Ann Intern Med 1999; 131: 348-351. [ Links ]
36. Ingenbleek Y, Barclay D, Dirren H. Nutritional significance in serum amino acid patterns in goitrous patients. Ann J Clin Nutr 1986; 43: 310-319. [ Links ]
37. Jacques PF, Selhub J, Bostom AG, Wilson PWF, Rosenberg IH. The effect of folic acid fortification on plasma folate and total homocysteine concentrations. N Engl J Med 1999; 340: 1449-1454. [ Links ]
38. Konecky N, Malinow MR, Tunick PA, et al. Correlation between plasma homocyst(e)ine and aortic atherosclerosis. Am Heart J 1997; 133: 534-40. [ Links ]
39. Lentz SR, Sadler JE. Inhibition of thrombomodulin surface expression and protein C activation by the thrombogenic agent homocysteine. J Clin Invest 1991; 88: 1906-14. [ Links ]
40. Loscalzo J. The oxidant stress of hyperhomocysteinemia. J Clin Invest 1996; 98: 5-7. [ Links ]
41. Malinow MR, Duell PB, Hess DL, et al. Reduction of plasma homocyst(e)ine levels by breakfast cereal fortified with folic acid in patients with coronary heart disease. N Engl J Med 1998; 338: 1009-15. [ Links ]
42. Mandel H, Brenner B, Berant M, et al. Coexistence of hereditary homocystinuria and factor V Leiden - effect on thrombosis. N Engl J Med 1996; 334: 763-8. [ Links ]
43. Mayer EL, Jacobsen Dw, Robinson K. Homocysteine and coronary atherosclerosis. J Am Coll Cardiol 1996; 27: 517-27. [ Links ]
44. McCully KS. La revolución de la homocisteína: la medicina en el nuevo milenio, I y II. Cardiovascular 1999; 20: 1-17. [ Links ]
45. McCully KS. Homocysteine and vascular disease. Nature Med 1996; 2: 386-389. [ Links ]
46. McCully KS. Macromolecular basis for homocysteine-induced changes in proteoglycan structure in growth and arteriosclerosis. Am J Pathol 1972; 66: 83-95. [ Links ]
47. McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of atherosclerosis. Am J Pathol 1969; 56: 111-28. [ Links ]
48. McCully KS, Ragsdale BD. Production of arteriosclerosis by homocysteinemia. Am J Pathol 1970; 61: 1-11. [ Links ]
49. McCully KS, Wilson RB. Homocysteine theory of arteriosclerosis. Atherosclerosis 1975; 22: 215-227. [ Links ]
50. Di Minno G, Davi G, Margaglione M, et al. Abnormally high thromboxane biosynthesis in homozygous homocystinuris: evidence for platelet involvement and probucol-sensitive mechanism. J Clin Invest 1993; 92: 1400-6. [ Links ]
51. Misra HP. Generation of superoxide free radical during the autooxidation of thiols. J Biol Chem 1974; 249: 2151-5. [ Links ]
52. Montalescot G, Ankri A, Chadefaux-Vekemans B, et al. Plasma homocysteine and the extent of atherosclerosis in patients with coronary artery disease. Int J Cardiol 1997; 60: 295-300. [ Links ]
53. Mudd SH, Skovby F, Levy LH, et al. The natural history of homocystinuria due to cystathionine beta synthase deficience. Am J Hum Genet 1985; 37: 1-31. [ Links ]
54. Naruszewicz M, Mirkiewicz E, Olszewski AJ, et al. Thiolation of low-density lipoprotein by homocysteine thiolactone causes increased aggregation and altered interaction with cultured macrophages. Nutr Metab Cardiovasc Dis 1994; 4: 70-77. [ Links ]
55. Nordström M, Kjellstrom T. Age dependency of cystathionine beta-synthase activity in human fibroblasts in homocysteinemia and atherosclerotic vascular disease. Atherosclerosis 1992; 94; 213-222. [ Links ]
56. Nygard O, Nordrehaug JE, Refsum H, Ueland PM, Farstad M, Vollset SE. Plasma homocysteine levels and mortality in patients with coronary artery disease. N Engl J Med, 1997; 337: 230-6. [ Links ]
57. Nygard O, Vollset SE, Refsum H, et al. Total plasma homocysteine and cardiovascular risk profile: the Hordaland Homocysteine Study. JAMA 1995; 274: 1526-33. [ Links ]
58. Pancharuniti N, Lewis CA, Sauberlich HE, et al. Plasma homocyst(e)ine, folate, and vitamin B12 concentrations and risk for early-onset coronary artery disease. Am J Clin Nutr 1994; 59: 940-8. [ Links ]
59. Parthasaraty S. Oxidation of low-density lipoprotein thiol compounds leads to its recognition by the acetyl LDL receptor. Biochim Biophys Acta 1987; 917: 337-40. [ Links ]
60. Perry IJ, Refsum H, Morris RW, Ebrahim SB, Ueland PM, Shaper AG. Prospective study of serum total homocysteine concentration and risk of stroke in middle-aged British men. Lancet 1995; 346: 1395-8. [ Links ]
61. Petri M, Roubenoff R, Dallal GE, Nadeau MR, Selhub J, Rosenberg IH. Plasma homocysteine as a risk factor for atherothrombotic events in systemic lupus erythematosus. Lancet 1996; 348: 1120-4. [ Links ]
62. Ratnoff OD. Activation of Hageman factor by L-homocystine. Science 1968; 162: 1007-9. [ Links ]
63. Refsum H, Wesenberg F, Ueland PM. Plasma homocysteine in children with acute lymphoblastic leukemia: changes during a chemotherapeitic regimen including methotrexate. Cancer Res 1991; 51: 828-35. [ Links ]
64. Ridker PM, Hennekens CH, Selhub J, Miletich JP, Malinow MR, Stampfer MJ. Interrelation of hyperhomocysteinemia, factor V Leiden, and risk of future venous thromboembolism. Circulation 1997; 95: 1777-1782. [ Links ]
65. Rodgers GM, Conn MT. Homocysteine, an atherogenic stimulus, reduces protein C activation by arterial and venous endothelial cells. Blood 1990; 75: 895-901. [ Links ]
66. Rodgers GM, Kane WH. Activation of endogenous factor V by a homocysteine-induced vascular endothelial cell activator. J Clin Invest 1986; 77: 1909-16. [ Links ]
67. Rosén SB, Sturk A: Activated protein C resistance: a major risk factor for thrombosis. Eur J Clin Chem Clin Biochem 1997; 35: 501-516. [ Links ]
68. Rowley DA, Halliwell B. Superoxide-dependent formation of hydroxil radicals in the presence of thiol compounds. FEBS Lett 1982; 138: 33-6. [ Links ]
69. Saltzman E, Mason JB, Jacques PF, et al. B vitamin supplementation lowers homocysteine levels in heart disease. Clin Res 1994; 42: 172A. [ Links ]
70. Selhub J, Jacques PF, Bostom AG, et al. Association between plasma homocysteine concentrations and extracranial carotid-artery stenosis. N Engl J Med 1995; 332: 286-91. [ Links ]
71. Selhub J, Jacques PF, Wilson PW, Rush D, Rosenberg IH. Vitamin status and intake as primary determinants of homocysteinemia in an elderly population. JAMA 1993; 270: 2693-8. [ Links ]
72. Stampfer MJ, Malinow MR, Willett WC, et al. A prospective study of plasma homocysteine and risk of myocardial infarction in US Physicians. JAMA 1992; 268: 877-81. [ Links ]
73. Starkebaum G, Harlan JM. Endothelial cell injury due to copper-catalyzed hydrogen peroxide generation from homocysteine. J Clin Invest 1986; 77: 1370-6. [ Links ]
74. de Stefano V, Martinelli I, Manucci PM, et al. The risk of recurrent deep venous thrombosis among heterozygous carriers of both factor V Leiden and the G20210A prothrombin mutation. N Engl J Med 1999; 341: 801-6. [ Links ]
75. Stehouwer CD, Weigenberg MP, van den Berg M, Jakobs C, Feskens EJ, Kromhout D. Serum homocysteine and risk of coronary heart disease and cerebrovascular disease in elderly men: a 10-year follow-up. Arterioscler Thromb Vasc Biol 1998; 18: 1895-901. [ Links ]
76. Tsai JC, Perrella MA, Yoshizumi M, et al. Promotion of vascular smooth muscle cell growth by homocysteine: a link to atherosclerosis. Proc Natl Acad Sci USA 1994; 91: 6369-73. [ Links ]
77. Tsai JC, Wang H, Perrella Ma, et al. Induction of cyclin A gene expression by homocysteine in vascular smooth muscle cells. J Clin Invest 1996; 97: 146-53. [ Links ]
78. Vermaak WJ, Ubbink JB, Barnars HC, Potgieter GM, van Jaarsveld H, Groenewald AJ. Vitamin B6 nutrition status and cigarette smoking. Am J Clin Nutr 1990; 51: 1058-61. [ Links ]
79. Ubbink JB, van der Merwe A, Delport A, et al. The effect of a subnormal vitamin B6 status on homocysteine metabolism. J Clin Invest 1996; 98: 177-84. [ Links ]
80. Ubbink JB, Vermaak WJ, Delport R, van der Merwe A, Becker PJ, Potgieter H. Effective homocysteine metabolism may protect South African blacks against coronary heart disease. Am J Clin Nutr 1995; 62: 802-8. [ Links ]
81. Ubbink JB, Vermaak WJ, van der Merwe A, Becker PJ. Vitamin B-12, vitamin B-6, and folate nutritional status in men with hyperhomocysteinemia. Am J Clin Nutr 1993; 57: 47-53. [ Links ]
82. Ueland PM ,Refsum H. Plasma homocysteine, a risk factor for vascular disease: plasma levels in health, disease, and drug therapy. J Lab Clin Med 1989; 114: 473-501. [ Links ]
83. Verhoef P, Hennekens CH, Allen RH, Stabler SP, Willett WC, Stampfer MJ. Plasma total homocysteine and risk of angina pectoris with subsequent coronary artery bypass surgery. Am J Cardiol 1997; 79: 799-801. [ Links ]
84. Verhoef P, Hennekens CH, Malinow MR, Kok FJ, Willett WC, Stampfer MJ. A prospective study of plasma homocyst(e)ine and risk of ischemic stroke. Stroke 1994; 25: 1924-30. [ Links ]
85. Verhoef P, Kok FJ, Kruyssen DA, et al. Plasma total homocysteine, B vitamins, and risk of coronary atherosclerosis. Arterioscler Thromb Vasc Biol 1997; 17: 989-95. [ Links ]
86. Wald NJ, Watt HC, Law MR, Weir DG, McPartlin J, Scott JM. Homocysteine and ischemic heart disease: results of a prospective study with implications regarding prevention. Arch Intern Med 1998; 158: 862-7. [ Links ]
87. Wall RT, Harlan JM, Harker LA, Striker GE. Homocysteine-induced endothelial cell injury in vitro: a model for the study of vascular injury. Thromb Res 1980; 18: 113-21. [ Links ]
88. Welch GN, Loscalzo J. Homocysteine and atherothrombosis. N Engl J Med 1998; 338: 1042-1050. [ Links ]
89. Welch GN, Upchurch GR, Loscalzo J. Hyperhomocyst(e)inemia and atherotrombosis. Ann N Y Acad Sci 1997; 811: 48-58. [ Links ]
90. Wilcken DE, Gupta VJ. Sulphur containing amino acids in chronic renal failure with particular reference to homocystine and cysteine-homocysteine mixed disulphide. Eur J Clin Invest 1979; 9: 301-7. [ Links ]

