










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.21 no.7 jul. 2004
| Estudio de los valores plasmáticos de Lp(a) en el defecto familiar de I. EJARQUE, J. T. REAL, J. F. ASCASO, F. J. CHAVES, E. MILIAN, M. A. PRIEGO, R, CARMENA Servicio de Endocrinología y Nutrición. Hospital Clínico Universitario. Departamento de Medicina.
|
| PLASMA LIPOPROTEIN A VALUIES IN FAMILIAL LIGAND DEFECTIVE APO B 100 IN A SOUTH EUROPEAN POPÙLATION
| ||
| RESUMEN Objetivo: Estudiar los valores plasmáticos de Lp(a) en el defecto familiar de unión de apo B 100 (DFB) en poblaciones sur Europea donde no existen datos al respecto. Métodos: Hemos estudiado a 19 heterocigotos DFB (8 varones), portadores de la mutación R3500Q del gen de la apo B y a 90 controles (34 varones). El diagnóstico genético se realizó por técnica de PCR-SSCP y secuenciación automática. En todos los sujetos se determinó de forma estandarizada las concentraciones plasmáticas de lípidos, apolipoproteína B y Lp(a). Resultados: Los valores plasmáticos de Lp(a) y su transformación logarítmica fueron significativamente mayores en el grupo con DFB frente al grupo control. Además, la prevalencia de Lp(a) > de 30 mg/dl, como punto de corte de alto riesgo para cardiopatía isquemia fue significativamente mayor en el grupo portador de la mutación R3500Q. Conclusión: Los sujetos portadores de la mutación R3500Q del gen de apo B mostraron valores plasmáticos superiores de Lp(a) que los controles, sin que conozcamos por el momento el mecanismo y sus implicaciones clínicas. PALABRAS CLAVE: Defecto familiar de unión de apo B 100. Lipoproteína A. Fenotipo lipoproteico. | ABSTRACT Aims: 1) to study lipoprotein (a) (Lp(a)) plasma values in subjects with familial ligand-defective apo B 100 (FDB) . Methods: We studied 19 heterozygous FDB subjects (8 males) from 12 families, carriers of R3500Q mutation on apo B gene and 90 controls (34 males). The genetic diagnosis was established with PCR-SSCP analysis and automatic sequencing. In all subjects plasma lipids, apolipoprotein B and Lp(a) levels were determined with standard procedures. Results: Subjects carriers of R3500Q mutation on apo B gene have significantly higher plasma Lp(a) and log transformed Lp(a) values and prevalence of Lp(a) > 30 cut point for coronary heart disease than controls. Conclusions: Subjects with FDB showed higher Lp(a) plasma values than controls, although the mechanism and the clinical consequences of these result are not known. KEY WORDS: Familial ligand defective apo B. Lipoprotein A, Lipid and lipoproteins. |
Ejarque I, Real JT, Ascaso JF, Chaves FJ, Milian E, Priego MA, Carmena R. Estudio de los valores plasmáticos de Lp(a) en el defecto familiar de unión de la apo B 100 en una población mediterránea del sur de Europa. An Med Interna (Madrid) 2004; 21: 322-325.
Trabajo aceptado: 11 de marzo de 2004
Correspondencia: Rafael Carmena, Departamento de Medicina. Hospital Clínico Universitario. Avda. Blasco Ibáñez, 17, E. 46010 Valencia. e-mail:carmena@uv.es
Este trabajo ha sido financiado por el Fondo de Investigaciones Sanitarias proyecto FIS 01/0056-02, Generalitat Valenciana proyecto GV01-46 y Red de Centros de Metabolismo y Nutrición del Instituto de Salud Carlos III (C 03/08).
INTRODUCCIÓN
El defecto familiar de unión de apo B 100 (DFB) se hereda de forma autosómica dominante y se caracteriza por la presencia de xantomas tendinosos, concentraciones plasmáticas elevadas de colesterol total (CT) y colesterol de la lipoproteína de baja densidad (LDL), y cardiopatía isquémica (CI) precoz (1,2). Cuatro mutaciones localizadas en el gen de la apo B (R3500Q, R3500W, R3531C y R3480P) son las responsables del DFB. La más prevalente de ellas es la mutación R3500Q (2-5).
La prevalencia estimada del DFB en nuestro país es muy baja, 2,8 x 10-5 para población general y aproximadamente del 1% en sujetos con diagnóstico clínico de HF, siendo más elevada en poblaciones centroeuropeas y norteamericanas (2,5-8). Nuestro grupo encontró y caracterizó en 1999 la primera familia española afectada de DFB (6) y el primer homocigoto (8). El despistaje de portadores de la mutación R3500Q en la zona donde se encontró la primera familia ha permitido detectar un mayor número de casos y conocer las características clínico - biológicas de la enfermedad en una zona del sur de Europa donde existen pocos datos sobre la misma.
La lipoproteína a (Lp(a)) es una lipoproteína LDL modificada, al unirse una apoproteína (a) por puentes disulfuro con la apoproteína B de la LDL (9). En múltiples estudios epidemiológicos, se ha demostrado que las elevaciones plasmáticas de la Lp(a) son un factor de riesgo cardiovascular (10). La Lp(a) es en parte aclarada del plasma por el receptor LDL (9). En sujetos con DFB se ha demostrado que la Lp(a) contiene apo B defectuosa, lo que podría contribuir a la elevación plasmática de esta partícula (11), al disminuir su aclaramiento plasmático por la falta de unión de la apo B defectuosa con el receptor de LDL.
Nuestro objetivo ha sido estudiar los valores plasmáticos de Lp(a) en el DFB en una población del sur de Europea donde no existen datos al respecto.
MATERIAL Y MÉTODO
SUJETOS
Hemos estudiado 19 heterocigotos DFB (8 varones) portadores de la mutación R3500Q del gen de la apo B 100 y 90 controles (34 varones). Los 19 heterocigotos DFB pertenecen a 12 familias relacionadas con la primera familia afectada por DFB descrita en nuestro país (7). Los 90 controles proceden de una selección aleatoria de sujetos sanos, sin antecedentes personales ni familiares de enfermedad coronaria, ni dislipemia. Estos sujetos sanos proceden de trabajadores, estudiantes, donantes de plasma y sangre del Banco de Sangre de nuestro Centro previamente seleccionados y reclutados por nuestro grupo de investigación.
Todos los participantes son caucasianos y residen en la Comunidad Autónoma de Valencia. El comité de ética de nuestro Centro aprobó el protocolo de estudio y todos los sujetos dieron su consentimiento por escrito, para participar en el mismo.
En todos ellos se realizó una historia clínica y exploración física completas. El índice de masa corporal (IMC) se calculó dividiendo el peso entre la talla al cuadrado (kg/m2). La cardiopatía isquémica se definió por una historia clínica documentada de infarto agudo de miocardio (clínica, elevación de CPK-MB y ECG patológico) o presencia de angor pectoris con test de esfuerzo y coronariografía patológicas.
MÉTODOS DE LABORATORIO
Medición de lípidos y apo B
Tras 12-14 horas de ayuno y en situación basal se recogió una muestra de sangre de la vena antecubital en tubos con EDTA (sistema Vacutainer) siendo centrifugadas en menos de 4 horas. Con situación basal nos referimos a la suspensión de fármacos hipolipemiantes 6 semanas previas a la toma de muestras para el estudio.
El colesterol y los triglicéridos (TG) fueron medidos por técnicas enzimáticas (12). El colesterol HDL (cHDL) se determinó tras precipitación de lipoproteínas con apo B con polianiones (13) el colesterol VLDL (cVLDL) tras separación de VLDL (d < 1.006 g/mL) por ultracentrifugación (14). El colesterol LDL (cLDL) se calculó por diferencia. Los niveles plasmáticos totales de apo B se midieron por inmunoturbimetría (15). La Lp(a) se midió por ELISA con un kit comercial (Macra Lp(a) SDI strategic diagnosis).
Los coeficientes de variación para los lípidos y lipoproteínas de nuestro laboratorio son < 5%.
MÉTODOS GENÉTICOS
La extracción de ADN se realizó con un método estandarizado (16). El diagnóstico genético de DFB se basó en análisis de PCR-SSCP para despistaje de la presencia de mutaciones localizadas en el gen de apo B responsables del DFB (R3480P, R3500Q, R3500W y R3531C). Posteriormente se llevó a cabo la secuenciación. El protocolo escalonado de diagnóstico molecular para hipercolesterolemias primarias que se realiza en nuestro centro se detalla en las referencias 17 y 18.
ANÁLISIS ESTADÍSTICO
Los datos fueron analizados utilizando el "Statistical Package for the Social Sciences (SPSS versión 9 para Windows)" y se expresan como medias, mediana y desviación estándar. Las medias de las variables cuantitativas se compararon utilizando el test de Mann Whitney. Las proporciones se compararon utilizando tablas de contingencia y Chi cuadrado (χ2) o el test de Fischer (n < 5).
Utilizamos el análisis de correlación de Pearson para analizar las asociaciones entre edad, IMC, lípidos y Lp(a) plasmáticos.
RESULTADOS
Despistaje de la mutación R3500Q en una zona de la Comunidad Valenciana
Partiendo de la primera familia diagnosticada de DFB en nuestro país y de la familia del homocigoto (6,8) se realizó un despistaje de la mutación R3500Q en todos los familiares accesibles relacionados.
Logramos contactar y estudiar la presencia o no de la mutación R3500Q en 51 sujetos, pertenecientes a 12 familias relacionadas. 19 de los 51 sujetos resultaron ser portadores de la mutación R3500Q.
CARACTERÍSTICAS BIOQUÍMICAS DE LOS SUJETOS DFB
Las características clínicas y concentraciones de lípidos y apoproteínas estudiados se presentan en la tabla I. No encontramos diferencias estadísticamente significativas en los parámetros referidos entre varones y mujeres portadores de la mutación R3500Q (datos no mostrados). En el grupo completo, no hallamos correlaciones entre la edad o el IMC y los lípidos y apoproteínas.
Los portadores de la mutación R3500Q presentaron de forma estadísticamente significativa mayores concentraciones plasmáticas de CT, cLDL y apo B comparados con el grupo control (Tabla I). Estas diferencias se mantuvieron al realizar la comparación entre géneros.
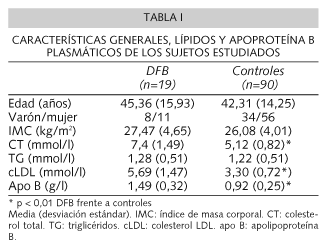
No hallamos diferencias estadísticamente significativas en el IMC, concentraciones plasmáticas de TG o cVLDL entre controles y portadores de la mutación R3500Q.
Al compararlos con el grupo control, los valores plasmáticos de Lp(a) y su transformación logarítmica fueron significativamente mayores en el grupo con DFB (Tabla II). Además, la prevalencia de Lp(a) > de 30 mg/dl, como punto de corte de alto riesgo para cardiopatía isquemia fue significativamente mayor en el grupo portador de la mutación R3500Q (28% frente a 8% respectivamente).

DISCUSIÓN
En nuestro estudio queda demostrado que los sujetos portadores de la mutación R3500Q del gen de la apo B presentan una elevación significativa de los valores plasmáticos de la Lp(a). Estudios anteriores, realizados en población holandesa y del Reino Unido, también observaron en los sujetos DFB una elevación plasmática de la Lp(a), comparado con los valores presentados por sus familiares no afectados. En el estudio de Perombelon y cols. (11) los sujetos portadores de la mutación R3500Q presentaban mayores valores de Lp(a) plasmática y menor tamaño de apo a, aunque existía una gran variabilidad en el fenotipo de apo a. En 5 sujetos DFB apareados por tamaño de apo a con sus familiares de primer grado no afectados también se demostró una mayor elevación plasmática de la Lp(a).
En el estudio de van der Hoek y cols. (19), la mediana de la Lp(a) plasmática no fue diferente entre los 85 sujetos DFB y sus 106 familiares DFB no afectados, sin embargo, al analizar parejas de hermanos sí se encontraron diferencias significativas, siendo los valores plasmáticos de la Lp(a) mayores en los portadores de la mutación R3500Q.
La elevación plasmática de la Lp(a) en sujetos DFB ha sido relacionada con la disminución del aclaramiento de las partículas de Lp(a) portadoras de la apoB defectuosa.
Por otra parte, diferentes estudios han demostrado que la Lp(a) plasmática también se encuentra elevada en sujetos con hipercolesterolemia familiar (HF) (20,21). Además, esta elevación ha sido relacionada con el papel que juega el receptor de LDL en el aclaramiento de la Lp(a) plasmática (9). Así, los sujetos HF con diferentes defectos en el receptor de LDL, deberían presentar un aclaramiento alterado de la Lp(a) plasmática, lo que justificaría la consiguiente elevación plasmática de la partícula. De forma contraria a esta hipótesis, un estudio en homocigotos HF con nula actividad del receptor de LDL mostraba que los valores plasmáticos de Lp(a) no dependían del aclaramiento de dicha partícula por el receptor de LDL (22). El mismo estudio mostraba también que los valores plasmáticos de Lp(a) dependían más de la síntesis de Lp(a) que de su catabolismo (22). Por otro lado, si bien el aclaramiento de la Lp(a) es posible por vía del receptor de LDL, este aclaramiento es mínimo por la gran afinidad de la partícula LDL por el mismo, que compite con la partícula de Lp(a) (23). Además, estudios de intervención con estatinas que inducen la sobreexpresión del receptor de LDL no demuestran disminuciones de la Lp(a) (24). Finalmente, en estudios de heterocigotos HF genéticamente caracterizados y de sus familiares de primer grado las elevaciones plasmáticas de la Lp(a) no se relacionaban con el tipo de mutación del receptor de LDL ni con la condición de HF (25,26).
Por tanto, la hipótesis de que la interacción de la apo B defectuosa que portan las partículas de Lp(a) de los sujetos con DFB es la causa de las elevaciones plasmáticas de Lp(a) encontradas en diferentes poblaciones estudiadas, al igual que en la nuestra, es probablemente falsa. De hecho, en el estudio de Perombelon y cols. (11), se demostró que las partículas de Lp(a) contenían una menor proporción de apo B defectuosa que las LDL de los sujetos DFB, indicando que la contribución de la interacción de la apoB defectuosa con el receptor de LDL en la elevación plasmática de la Lp(a) era pequeña y no justificaba la elevación plasmática de la Lp(a) en estos sujetos. Por otro lado y en este sentido, estudios in vivo del metabolismo de la Lp(a) demuestran que la fracción catabólica de la apo B y apo a contenidas en la Lp(a) son similares pero un 50% menores a la fracción catabólica observada para la LDL (9).
Es pues necesario realizar un mayor número de estudios in vivo del metabolismo de la Lp(a) en sujetos DFB para poder conocer la causa de la elevación de esta partícula en estos sujetos.
Si bien la Lp(a) plasmática se encuentra elevada en pacientes afectados del DFB, no se han realizado estudios para conocer el impacto de esta elevación en el riesgo cardiovascular de estos sujetos, teniendo en cuenta que en otras poblaciones la Lp(a) es un reconocido factor de riesgo cardiovascular (10).
La mayor limitación de nuestro estudio, es el bajo número de sujetos con DFB incluidos, lo que implica un elevado error beta y la relativa homogeneidad molecular por el vínculo familiar existente entre los sujetos con DFB. Esta relativa homogeneidad podría influir en la expresión del fenotipo lipoproteico.
En conclusión, la Lp(a) plasmática se encuentra elevada en sujetos con DFB. Es necesario realizar estudios del metabolismo de la Lp(a) en sujetos portadores de la mutación R3500Q para aclarar el papel que juega la apoB defectuosa y el receptor de LDL en el aclaramiento de la partícula Lp(a). Además, también es necesario aclarar si esta elevación de la Lp(a) plasmática es un factor de riesgo cardiovascular adicional en los sujetos con DFB.
Bibliografía
1. Myant NB. Familial defective apolipoprotein B-100: a review, including some comparison with familial hypercholesterolemia. Atherosclerosis 1993; 104: 1-18. [ Links ]
2. Rauh G, Séller C, Schuster H, Wolfgram G, Zöllnner N. Familial defective apolipoprotien B-100: a common cause of primary hyperlipemia. Clin Investigator 1995; 95: 1225-1234. [ Links ]
3. Pullinger CR, Hennessy LK, Chatterton JE, et al. Familial ligand-defective apolipoprotein B: identification of a new mutation that decreases LDL receptor binding affinity. J Clin Invest 1995; 95: 1225-1234. [ Links ]
4. Gaffney D, Reid JM, Cameron IM, et al. Independent mutations at codon 3500 of the apolipoprotein B gene are associated with hyperlipidaemia. Arterioscle Thromb Vasc Biol 1995; 15: 1025-1029. [ Links ]
5. Miserez AR, Keller U. Differences in the phenotypic characteristics of subjects with familial defective apolipoprotein B-100 and familial hypercholesterolemia. ATVB 1995; 15: 1719-1729. [ Links ]
6. Real JT, Chaves FJ, Ascaso JF, Armengod ME, Carmena R. Estudio del defecto familiar de la apo B-100 en sujetos con el diagnóstico clínico de hipercolesterolemia primaria: identificación de la primera familia afectada en España. Med Clin (Barc) 1999; 113: 15-17. [ Links ]
7. Castillo S, Tejedor D, Mozas P, Reyes G, Civeira F, Alonso R, Ros E, Pocovi M, Mata P. Atherosclerosis 2002; 165: 127-135. [ Links ]
8. Real JT, Chaves JF, Martín de Llano JJ, et al. Identificación y caracterización del primer homocigoto español con defecto familiar de unión de la apolipoproteína B. Med Clin (Barc) 2001; 116: 138-141. [ Links ]
9. Demant T, Seeberg K, Bedynek A, Seidel D. The metabolism of lipoprotein(a) and other apolipoprotein B-containing lipoproteins: a kinetic study in humans. Atherosclerosis 2001; 157: 325-39. [ Links ]
10. Pedro- Botet J, Rubies-Prat J. Lipoproteína (a) y aterotrombosis. Clin Invest Arterioscler 2003; 15: 258-260. [ Links ]
11. Perombelon YF, Gallagher JJ, Myant NB, Soutar AK, Knight BL. Lipoprotein(a) in subjects with familial defective apolipoprotein B100. Atherosclerosis 1992; 92: 203-12. [ Links ]
12. Allain CC, Poon LS, Chan CSG, Richmond W, Fu PC. Enzymatic determination of total serum cholesterol. Clin Chem 1974; 20: 470-5. [ Links ]
13. Burstein M, Scholnick HR, Morfin R. Rapid method for the isolation of lipoproteins from human serum by precipitation with polyanions. J Lipid Res 1970; 11: 583-95. [ Links ]
14. Havel RJ, Eder HJ, Bragdon JH. The distribution and chemical composition of centrifugally separated lipoproteins in human serum. Eur J Clin Invest 1995; 34: 1345-54. [ Links ]
15. Rosseneu M, Vercaemst R, Steinberg KK, Cooper GR. Some considerations of methodology and standarization of apolipoprotein B immunoassays. Clin Chem 1983; 29: 427-33. [ Links ]
16. Tilzer L, Thomas S, Moreno RF. Use of silica gel polymer for DNA extraction with organic solvents. Anal Biochem 1989; 183: 13-15. [ Links ]
17. García-García AB, Real JT, Puig O, et al. Molecular genetics of familial hipercolesterolemia in Spain: ten novel LDLR mutations and population analysis. Hum Mutat 2001; 454: 1-9. [ Links ]
18. Chaves FJ, Real JT, García-García AB, et al. Genetic diagnosis of familial hypercholesterolemia in a south European outbreed population: influence of low density lipoprotein (LDL) receptor gene mutations on treatment response to simvastatin in total, LDL and high density lipoprotein cholesterol. J Clin Endocrinol Metab 2001; 86: 4926-4932. [ Links ]
19. van der Hoek YY, Lingenhel A, Kraft HG, Defesche JC, Kastelein JJ, Utermann G. Sib-pair analysis detects elevated Lp(a) levels and large variation of Lp(a) concentration in subjects with familial defective ApoB. J Clin Invest 1997; 99: 2269-73 [ Links ]
20. Seed M, Hopplicher F, Reaveley D, Mc Carthy S, Thompson G, Boerwinkle E, Uterman G. Relation of serum lipoprotein (a) concentration and apolipoprotein a phenotype to coronary heart disease in patients with familial hypercholesterolemia. N Engl J Med 1990; 322: 1494-1499. [ Links ]
21. Kraft HG, Lingenhel A, Raal FJ, Hohenegger M, Utermann G. Lipoprotein(a) in homozygous familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2000; 20: 522-8. [ Links ]
22. Rader DJ, Mann WA, Cain W, Kraft HG, Usher D, Zech La, et al. The low density lipoprotein receptor is not required for normal catabolism of Lp(a) in humans. J Clin Invest 1995; 95: 1403-1408. [ Links ]
23. Armstrong VW, Walli AK, Seidel D. Isolation characterization and uptake in human fibroblasts of an apo(a) free lipoprotein obtained on reduction of lipoprotein(a). J Lipid Res 1985; 26: 1314-1318. [ Links ]
24. Thiery J, Armstrong VW, Schleef J, Creutzfeld C, Creutzfeld W, Seidel D. Serum lipoprotein Lp(a) concentrations are not influence by an HMG CoA reductase inhibitor. Klin Wochenschr 1988; 66: 462-466. [ Links ]
25. Carmena R, Lussier-Cacan S, Roy M, Minnich A, Lingenhel A, Kronenberg F, Davignon J. Lp(a) levels and atherosclerotic vascular disease in a sample of patients with familial hypercholesterolemia sharing the same gene defect. Arterioscler Thromb Vasc Biol 1996; 16: 129-36. [ Links ]
26. Real JT, Ascaso JF, Chaves FJ, Tenes S, Priego MA, Puig O, Armengod ME, Carmena R. Plasma Lp(a) values in familial hypercholesterolemia and its relation to coronary heart disease. Nutr Metab Cardiovasc Dis 1999; 941-4. [ Links ]

