










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.22 no.3 mar. 2005
| Síndrome "overlap" hepatitis autoinmune-cirrosis biliar primaria. Aportación de dos nuevos casos F. J. FERNÁNDEZ FERNÁNDEZ, J. DE LA FUENTE AGUADO, S. PÉREZ FERNÁNDEZ, B. SOPEÑA PÉREZ-ARGÜELLES, A. NODAR GERMIÑAS, M. BUTRÓN VILA1 Servicio de Medicina Interna. 1Servicio de Anatomía Patológica. Complexo Hospitalario Universitario Xeral-Cíes. Vigo
|
| PRIMARY BILIARY CIRRHOSIS-AUTOIMMUNE HEPATITIS OVERLAP SYNDROME. CONTRIBUTION OF TWO NEW CASES
| ||
| RESUMEN El síndrome "overlap" hepatitis autoinmune (HAI)-cirrosis biliar primaria (CBP) se caracteriza por un solapamiento de hallazgos clínicos, analíticos, inmunológicos e histológicos tanto de HAI como de CBP, cuya patogenia y tratamiento más adecuado se desconoce en la actualidad. Describimos dos nuevos casos de esta entidad, que debutaron con un brote de hepatitis aguda colestásica acompañado de hipergammaglobulinemia. En el primer caso se demostró la presencia de los anticuerpos AMA, ASMA y anti-LKM1; y en el segundo de los ANA. Los hallazgos histológicos mostraron alteraciones sugestivas de HAI y de CBP. Tras la instauración de tratamiento inmunosupresor, asociado a ácido ursodesoxicólico en un caso, se observó una buena evolución clínica. PALABRAS CLAVE: Overlap. Hepatitis autoinmune. Cirrosis biliar primaria. Tratamiento. | ABSTRACT The autoimmune hepatitis (AIH)-primary biliary cirrhosis (PBC) overlap syndrome is characterized for clinical, biochemical, immunological, and histological features overlapping those of AIH and PBC, whose pathogenesis and more appropriate treatment are unknown at present. We describe two new patients of this entity, which made debut with cholestasic acute hepatitis accompanied of hypergammaglobulinemia. In the first patient was demonstrated the presence of AMA, ASMA, and anti-LKM1 autoantibodies; and ANA in the second one. The histological findings showed changes suggestive of AIH and PBC. After the start of immunosuppressive treatment, associated to ursodeoxycholic acid in one patient, a successfull outcome was observed. KEY WORDS: Overlap. Autoimmune hepatitis. Primary biliary cirrhosis. Treatment. |
Fernández Fernández FJ, de la Fuente Aguado J, Pérez Fernández S, Sopeña Pérez-Argüelles B, Nodar Germiñas A, Butrón Vila M. Síndrome "overlap"hepatitis autoinmune-cirrosis biliar primaria. Aportación de dos nuevos casos. An Med Interna (Madrid) 2005; 22: 136-138.
Trabajo aceptado: 11 de noviembre de 2004
Correspondencia: Francisco Javier Fernández Fernández. Servicio de Medicina Interna. Complexo Hospitalario Universitario Xeral-Cíes. C/ Pizarro, 22. 36204 Vigo, Pontevedra. e-mail: fjfer@mundo-r.com
INTRODUCCIÓN
La hepatitis autoinmune (HAI) es una enfermedad crónica que se caracteriza por inflamación y necrosis hepatocelular en presencia de anomalías serológicas, y que en ocasiones se acompaña de manifestaciones extrahepáticas de autoinmunidad. Aproximadamente entre el 7 y el 10% de las hepatitis autoinmunes presentan características de cirrosis biliar primaria (CBP) (1,2). Esta asociación está ampliamente reconocida en la actualidad y se denomina síndrome "overlap" HAI-CBP (3). La diferente naturaleza de esta enfermedad, unido al desconocimiento que existe en cuanto a su patogenia, manejo terapéutico y evolución, nos anima a describir dos nuevos casos para su mejor comprensión.
CASOS APORTADOS
Caso 1: Mujer de 69 años sin enfermedades previas ni tratamientos habituales, que inició en septiembre de 1999 una clínica de 15 días de evolución consistente en ictericia progresiva, coluria, hipocolia, astenia y dolor epigástrico intermitente. En la exploración física se apreciaron como únicos datos reseñables ictericia mucocutánea y hepatomegalia. Las pruebas de imagen, ecografía y tomografía axial computadorizada (TAC) abdominal, sólo mostraron una hepatomegalia homogénea. En los análisis realizados destacaban transaminasa glutámico-oxalacética (GOT) 775 UI/L, transaminasa glutámico-pirúvica (GPT) 1116 UI/L, gammaglutamiltranspeptidasa (GGT) 121 UI/L, fosfatasa alcalina (FA) 362 UI/L, bilirrubina total 15,9 mg/dl (directa 6,4 mg/dl), anticuerpos antinucleares (ANA) positivos a un título de 1/640, e hipergammaglobulinemia policlonal con un valor de IgG de 2110 mg/dl. Los anticuerpos antimitocondriales (AMA), anticuerpos antimúsculo liso (ASMA), anticuerpos anticitoplasma de neutrófilo (ANCA) y anticuerpos antimicrosomales de hígado y riñón (anti-LKM1) resultaron negativos, así como las serologías frente a virus de hepatitis A (VHA), hepatitis B (VHB) y hepatitis C (VHC). El cuadro cedió completamente en un mes sin tratamiento, produciéndose la normalización de todos los parámetros previamente alterados. Cuatro meses después presentó un nuevo episodio de ictericia, coluria, acolia, astenia y dolor en hipocondrio derecho de 10 días de evolución. En las pruebas de imagen únicamente se apreció una hepatomegalia homogénea. Los parámetros del autoanalizador mostraron una GOT de 1665 UI/L, GPT de 1366 UI/L, GGT de 69 UI/L, FA de 380 UI/L y bilirrubina de 21.6 mg/dl. Los ANA fueron nuevamente positivos (a título de 1/320) con negatividad del resto de autoanticuerpos. Se practicó una biopsia que mostró cierto grado de distorsión de la arquitectura hepática con espacios porta ensanchados por infiltrado linfocitario, neutrófilos y escasos eosinófilos, con proliferación ductular periférica y rotura de la membrana limitante e inflamación de algunos ductos, apreciándose además lobulillos con hepatocitos con focos de hiperplasia y colestasis, alguno de ellos necrótico, con escasas necrosis focales en el parénquima (Fig. 1). Se instauró tratamiento con prednisona y azatioprina, que se mantuvo durante un año, obteniéndose la remisión completa, tanto clínica como analítica. Tras tres años de seguimiento la paciente permanece asintomática.

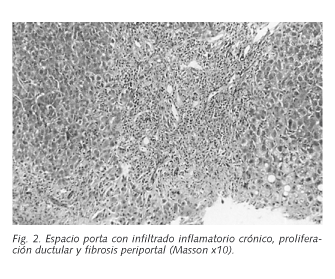
Caso 2: Mujer de 31 años, fumadora de 10 paquetes/año, a tratamiento con anticonceptivos orales de forma intermitente en los últimos 8 años hasta 1 año antes, que presentó en octubre de 1997 un cuadro de 1 mes de evolución de ictericia progresiva, náuseas, orinas colúricas y heces acólicas. En la exploración física se evidenció eritema palmar bilateral, ictericia y hepatomegalia. En las pruebas de imagen (ecografía y TAC abdominal) se puso de manifiesto una hepatomegalia homogénea. Los análisis practicados demostraron: GOT 384 UI/L, GPT 496 UI/L, GGT 78 UI/L, FA 553 UI/L y bilirrubina 11,5 mg/dl (directa 4,8 mg/dl), así como hipergammaglobulinemia policlonal, con 3030 mg/dl de IgG. Las serologías a VHA, VHB y VHC resultaron negativas. Los ANA fueron negativos. Los AMA resultaron positivos (1/1280), así como los ASMA (1/320), Anti-LKM1 (1/640) y ANCA (1/40). Se procedió a la realización de una biopsia hepática que mostró leve distorsión focal debido a bandas fibrosas porto-portales ocasionales, con infiltrado inflamatorio linfocitario en los espacios porta formando folículos linfoides, en ocasiones alrededor de los ductos biliares. Dicho infiltrado traspasaba los límites del espacio porta, observándose necrosis "piecemeal" moderada/severa. Se apreciaron hepatocitos alrededor del espacio porta con acúmulo de proteína transportadora de cobre, e incremento de la celularidad inflamatoria en el lobulillo, tanto en sinusoides como en el parénquima, con citolisis e incremento de la trama fibrosa intersticial (Fig. 2). Se inició tratamiento con 30 mg de prednisona con posterior reducción de su dosis, 50 mg de azatioprina y ácido ursodesoxicólico. Se produjo la resolución completa del cuadro clínico con normalización de los análisis, persistiendo discretamente elevadas, después de seis años de evolución, la FA y la GGT.
DISCUSIÓN
Aunque CBP y HAI son dos entidades nosológicas diferentes, en ocasiones el diagnóstico es difícil debido a la similitud de características clínicas correspondientes a ambas enfermedades. Aproximadamente entre el 7% y el 10% de las hepatitis autoinmunes tienen unas características clínicas e inmunológicas que remedan la CBP, denominándose en la actualidad a esta enfermedad como síndrome "overlap" CBP-HAI (1,2). Aunque reconocida desde hace más de 25 años (4), poco se ha investigado sobre la misma en cuanto a patogenia y tratamiento, resultando importante su diagnóstico y reconocimiento puesto que el tratamiento es diferente en cada caso. En este sentido se han intentado evaluar sistemas de puntuación para diferenciar las variantes de las enfermedades hepáticas autoinmunes, si bien todavía no han sido aceptados universalmente (5,6).
El síndrome "overlap" CBP-HAI no tiene unos criterios plenamente validados para su diagnóstico, aunque suelen utilizarse los descritos por Chazouillères y cols. (2), que requieren al menos dos criterios diagnósticos para cada una de las enfermedades, que comprenden para la CBP: a) FA al menos dos veces el límite superior del valor normal o GGT al menos cinco veces mayor al límite superior de la normalidad; b) presencia de AMA; y c) lesiones en los conductos biliares floridas; y para la HAI: a) niveles de GPT al menos cinco veces sobre el límite superior de la normalidad; b) valores de IgG al menos dos veces superiores al valor normal o positividad para los ASMA; y c) necrosis "piecemeal" linfocitaria periseptal o periportal severa. Lógicamente, debe excluirse la presencia de obstrucción biliar, así como confirmar la negatividad de las serologías virales y consumo de alcohol o fármacos hepatotóxicos.
Este síndrome también debe ser distinguido de la denominada colangitis autoinmune, que se caracteriza por hallazgos clínicos y patológicos concordantes con CBP pero con negatividad de los AMA, frecuente positividad para los ANA y/o ASMA, y presencia de cambios histológicos de colangitis no supurativa crónica (2,7). Actualmente esta entidad se considera una variante de la CBP, recomendándose para su tratamiento el ácido ursodesoxicólico, ya que los corticoides son generalmente ineficaces.
El síndrome "overlap" CBP-HAI suele afectar a mujeres, con una edad media de 50 años, y frecuentemente asocia enfermedades extrahepáticas, tales como síndrome de Sjögren, fenómeno de Raynaud o artropatías (1,2,8). La sintomatología clínica es inespecífica, y en los análisis de laboratorio se observa tanto un perfil de hepatitis como colestásico, si bien suele predominar este último. Los hallazgos anatomopatológicos revelan la presencia de una colangitis con un infiltrado inflamatorio de tipo linfoplasmocitario, que se acompaña en ocasiones de depósito de cobre hepático, así como la presencia de una hepatitis con necrosis "piecemeal" (3).
Se desconoce su evolución espontánea por lo que el tratamiento representa un dilema terapéutico. Para ello se ha ensayado el ácido ursodesoxicólico, el cual ha demostrado una menor eficacia que en la CBP, y los esteroides, con los que se ha objetivado una disminución significativa de los valores de FA, GPT e IgG sin llegar a una completa normalización (1,2,8-10). En los casos en que se han empleado conjuntamente se ha evidenciado una mejoría de los parámetros bioquímicos hepáticos (2,11,12). De todos modos todas estas estrategias son empíricas, y la recomendación actual es tratar las manifestaciones predominantes de la enfermedad.
Nosotros describimos dos nuevos casos de inicio agudo, definidos por la elevación de las enzimas hepáticas, tanto las propias de colestasis como las de hepatitis, los hallazgos histológicos compatibles, la presencia de hipergammaglobulinemia policlonal, y la positividad de los autoanticuerpos, en el primer caso de los ANA, y en el segundo de los AMA, ASMA y Anti-LKM1.
El primer caso presentó dos brotes de hepatitis aguda ictérica, iniciándose tras el segundo episodio tratamiento inmunosupresor (esteroides y azatioprina), alcanzando la remisión completa, sin evidencia de recidiva de la enfermedad en tres años de seguimiento. Por el contrario, en el segundo caso, debemos reseñar que con el uso de inmunosupresores y ácido ursodesoxicólico se consiguió una resolución del brote agudo, permaneciendo asintomática desde entonces, si bien persiste con un patrón analítico de leve colestasis.
En resumen, consideramos necesaria una descripción pormenorizada de esta condición, a fin de dilucidar su mecanismo patogénico, definir unos criterios diagnósticos y determinar las posibilidades terapéuticas.
Bibliografía
1. Czaja AJ. Frequency and nature of the variant syndromes of autoimmune liver disease. Hepatology 1998; 28: 360-365. [ Links ]
2. Chazouillères O, Wendum D, Serfaty L, Montembault S, Rosmorduc O, Poupon R. Primary biliary cirrhosis-autoimmune hepatitis overlap syndrome: clinical features and response to therapy. Hepatology 1998; 28: 296-301. [ Links ]
3. Czaja AJ. The variant forms of autoimmune hepatitis. Ann Intern Med 1996; 125: 588-598. [ Links ]
4. Klopell G, Seifert G, Lindner H, Dammerman R, Sack HJ, Berg PA. Histopathological features in mixed types of chronic aggressive hepatitis and primary biliary cirrhosis. Virchows Arch A Pathol Anat Histopathol 1977; 373: 143-160. [ Links ]
5. Talwalkar JA, Keach JC, Angulo P, Lindor KD. Overlap of autoimmune hepatitis and primary biliary cirrhosis: an evaluation of a modified scoring system. Am J Gastroenterol 2002; 97: 1191-1197. [ Links ]
6. Yamamoto K, Terada R, Okamoto R, Hiasa Y, Abe M, Onji M et al. A scoring system for primary biliary cirrosis and its application for variants forms of autoimmune liver disease. J Gastroenterol 2003; 38: 52-59. [ Links ]
7. Goodman ZD, McNally PR, Davis DR, Ishak KG. Autoimmune cholangitis: a variant of primary biliary cirrhosis. Clinico-pathologic and serologic correlations in 200 cases. Dig Dis Sci 1995; 40: 1232-1242. [ Links ]
8. Lohse AW, Meyer zum Büschenfelde KH, Franz B, Kanzler S, Gerken G, Dienes HP. Characterization of the overlap syndrome of primary biliary cirrhosis (PBC) and autoimmune hepatitis: evidence for it being a hepatitic form of PBC in genetically susceptible individuals. Hepatology 1999; 29: 1078-1084. [ Links ]
9. Joshi S, Cauch-Dudek K, Wanless IR, Lindor KD, Jorgensen R, Batts K et al. Primary biliary cirrhosis with additional features of autoimmune hepatitis: response to therapy with ursodeoxycholic acid. Hepatology 2002; 35: 409-413. [ Links ]
10. Ben-Ari Z, Dhillon AP, Sherlock S. Autoimmune cholangiopathy: part of the spectrum of autoimmune chronic active hepatitis. Hepatology 1993; 18: 10-15. [ Links ]
11. Perrillo RP, Mason AL, Jacob S, Gerber MA. Hepatitis and cholestasis in a middle-aged woman. Hepatology 1996; 24: 730-734. [ Links ]
12. Colombato LA, Álvarez F, Cote L, Huet PM. Autoimmune cholangiopathy: the result of consecutive primary biliary cirrosis and autoimmune hepatitis? Gastroenterology 1994; 107: 1839-1843. [ Links ]

