










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales de Medicina Interna
versión impresa ISSN 0212-7199
An. Med. Interna (Madrid) vol.25 no.3 mar. 2008
Mastocitosis sistémica. Revisión sistemática
Systemic mastocitosis. Systematic review
M.J. Molina-Garrido, A. Mora1, C. Guillén-Ponce, M. Guirado-Risueño, M.J. Molina, M.A. Molina, A. Carrato
Servicios de Oncología Médica y 1Medicina Interna. Hospital General Universitario. Elche. Alicante
Dirección para correspondencia
RESUMEN
La mastocitosis es una enfermedad definida por un crecimiento anormal y por la acumulación de mastocitos. En la clasificación de consenso de la OMS del 2001, se distinguió entre procesos bien limitados a piel (mastocitosis cutánea) o bien acumulados a nivel de otros tejidos: médula ósea y/u otros órganos extracutáneos (mastocitosis sistémica) como huesos, hígado, bazo o ganglios linfáticos (70% afectación ósea, con patrón osteolítico u osteoblástico, seguida de 50% de hepatoesplenomegalia).
La sintomatología más común en estos enfermos es la afectación de la piel por urticaria pigmentosa (más frecuente en la infancia) o telangiectasia macularis pertans (más frecuente en adultos) donde los mastocitos pueden estar recluidos mucho tiempo, con clínica que proviene de sus mediadores, siendo los niveles de triptasa el reflejo de la carga tumoral. El manejo de esta enfermedad se basa en la administración de tratamiento sintomático con antagonistas de histaminas H1 y H2, así como cromoglicato disódico, necesitando terapia citorreductora sólo en las variantes agresivas de mastocitos sistémica (asociadas a mutación del receptor de tirosin kinasa c-kit D816V) o asociadas a SMP (proliferación de mastocitos e hipereosinofilias asociado a la expresión del gen de fusión FIP1L1-PDGFRA).
El interferón tiene un efecto beneficioso sobre los síntomas dermatológicos, hematológicos, gastrointestinales y sistémicos, así como en los esqueléticos, debido a su capacidad de aumentar la densidad ósea y reducir los episodios dolorosos, siendo beneficioso el tratamiento inicial con prednisona.
Palabras clave: Mastocitosis sistémica. Niveles de triptasa. Lesiones óseas mixtas.
ABSTRACT
Mastocitosis is a hematologic malignance characterized by an anormal proliferation of mastocites. In a consess classification in 2001, it was distinguished between matters limited to skin and systemic matters (70% of osseous involvement and 50% of hepatomegaly).
The most typical symptoms are skin lesions and systemic manifestations due to mediators secreted by tumoral cells. They are useful chemotherapy to reduce the tumoral burden and antyhystaminic to control systemic manifestations.
Interpheron is useful in most of systemic and local manifestations, and it is recommended to use prednisona before the use of this medication.
Key words: Systemic mastocitosis. Triptase levels. Litic and blastic bone lesions.
Introducción
La mastocitosis sistémica (MS) es una enfermedad clonal de los progenitores mastocíticos de la médula ósea. El cuadro clínico en la MS varía desde una forma asintomática (indolente) a una forma altamente agresiva con una supervivencia muy corta (leucemia de mastocitos) (1).
En 1869, Nettleship describió la urticaria pigmentosa (UP) y en 1887, Unna describió un amento en el número de mastocitos en la UP. Fue ya en 1949, cuando Ellis describió una enfermedad sistémica asociada a hiperplasia de mastocitos.
La mayoría de los pacientes afectos son adultos, aunque la enfermedad puede ocurrir a cualquier edad. En función del momento de aparición de los síntomas, en especial, de las lesiones cutáneas, se define una mastocitosis de la infancia (antes de la pubertad) y una mastocitosis de la edad adulta (después de la pubertad). En un reducido número de casos, se puede detectar una historia familiar (mastocitosis familiar).
Los síntomas derivan de los productos secretados por los mastocitos o de la infiltración de órganos por los mismos. Datos recientes sugieren que la MS se comporta como una enfermedad mieloproliferativa que involucra a los progenitores hematopoyéticos pluripotenciales. Esta idea está potenciada por la existencia de mutaciones en c-kit-816 (2).
Es de especial importancia que se sospeche una mastocitosis sistémica en pacientes con inestabilidad vascular inexplicable, un shock anafiláctico sin causa conocida, enrojecimiento cutáneo y facial idiopático, diarrea, cefaleas y otros síntomas que podrían estar relacionados con la secreción de mediadores (3).
Principios básicos
Los mastocitos derivan de sus progenitores hematopoyéticos y, tanto en humanos, como en roedores, su principal factor de crecimiento es el factor de células stem (SCF) (4).
Los mastocitos que están destinados a residir en tejidos periféricos, como la piel, pulmón e intestino, se originan en la médula ósea y circulan en la sangre como células precursoras CD34+ que posteriormente atraviesan las células endoteliales para depositarse en los tejidos periféricos, donde se diferencian y maduran (5). Allí, forman gránulos de secreción, y expresan en su superficie otros receptores (FcεRI).
Son múltiples las citoquinas y los factores que contribuyen a la mastopoyesis. Una citoquina fundamental en este proceso es el factor de las células stem (CSF). Éste induce el desarrollo de los mastocitos a partir de sus progenitores y promueve su diferenciación terminal y su maduración (6). Los efectos de SCF sobre los mastocitos y sus progenitores está mediado a través de KIT, un receptor transmembrana del tipo de tirosin-kinasa codificado por el proto-oncogen c-kit (7). Los ratones de laboratorio que carecen de c-kit o del gen de SCF, carecen de mastocitos. Por todo esto, se sabe que SCF y KIT son unas moléculas críticas en la regulación del desarrollo de los mastocitos, su proliferación y su supervivencia. Por este motivo, las mutaciones que impliquen una ganancia de función en el gen c-kit se asocian con un mayor crecimiento de los mastocitos. Estas mutaciones, en especial, la c-kit D816V (Asp-816-Val), se identifican con frecuencia en los pacientes con mastocitosis sistémica (8-10).
Al contrario de lo que ocurre con los mastocitos normales, los mastocitos de las mastocitosis sistémicas normalmente expresan CD25 y CD2; las células más inmaduras también podrían expresar otros antígenos adicionales. Los progenitores más inmaduros (tanto los normales como los neoplásicos), expresan CD34 y CD13, así como CD117 (11).
Clasificación
Basándonos en los hallazgos clínicos y en los síntomas, se puede describir cuatro grandes grupos de pacientes con mastocitosis sistémica (Tabla I).
En la mayoría de los pacientes (80%), la mastocitosis sólo afecta a la piel. En el resto de los casos, puede estar infiltrada la médula ósea, el bazo, los ganglios o el tracto gastrointestinal.
En la mastocitosis de la médula ósea, un tipo especial de mastocitosis, la enfermedad está restringida a dicho órgano; es una forma indolente de mastocitosis sin lesiones cutáneas. En estos pacientes, los niveles de triptasa sérica son normales o muy bajos, en contraste a las mastocitosis malignas (12).
Fisiopatología y clínica
Los mastocitos y los basófilos tienen algunas características que los hacen similares, pero también presentan numerosas diferencias. Mientras que el CSF es el principal factor de crecimiento para los mastocitos, la interleukina 3 (IL-3) lo es para los basófilos humanos, que completan su maduración en la médula ósea y entran al torrente sanguíneo cuando ya tienen gránulos de secreción en su interior, receptores de superficie tipo FcεRI y una pequeña cantidad de receptores Kit. Estos circularán por sangre, y solo se depositarán a los tejidos que sufran un proceso inflamatorio (13).
En humanos, los gránulos de secreción de los mastocitos contienen altas cantidades de triptasa, histamina y heparina, quimasa y proteoglicanos de condroitín sulfato E. Una subpoblación de mastocitos también contiene cantidades altas de catepsina G y carboxipeptidasa. Aquellos mastocitos que sólo tienen triptasa se denominan células MCT.
Los basófilos contienen cantidades de histamina similares a la de los mastocitos. Sin embargo, a diferencia de estos, los basófilos contienen sulfato de condroitina tipo A en sus gránulos, así como proteínas eosinofílicas, pequeñas cantidades de triptasa y una proteína aún no bien caracterizada (14,15).
Estos criterios de distinción entre mastocitos y basófilos aún no están demasiado claros. Se ha aislado células metacromáticas (presumiblemente basófilos) de sangre periférica de pacientes con asma activa y con reacciones de hipersensibilidad que eran fuertemente positivas para triptasa, quimasa y Kit de superficie, lo que sugiere que los basófilos de la sangre periférica podrían adquirir patrones de mastocitos bajo determinadas situaciones clínicas (16).
La clínica de la mastocitosis sistémica (MS) se debe a que los tejidos están ocupados por la masa de mastocitos y estos reaccionan mediante fibrosis y liberación de sustancias activas que actúan a nivel local (urticaria pigmentosa, dolor abdominal cólico, gastritis, úlcera péptica) y a nivel sistémico (cefalea, prurito, colapso vascular o rubefacción). Estas manifestaciones clínicas pueden agravarse por la toma de AINEs, alcohol o narcóticos del tipo de la codeína, o durante una anestesia general (Fig. 1).
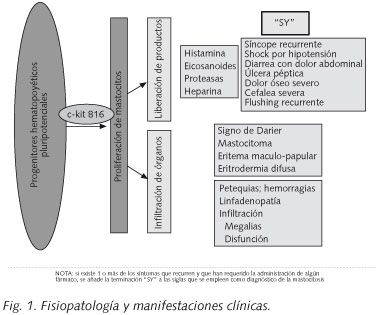
La urticaria pigmentosa es la manifestación cutánea más típica y más frecuente de la mastocitosis, tanto en niños como en adultos. Las lesiones son máculas de pequeño tamaño de color amarillento-rojizo. En casos más raros, pueden ser nodulares. La segunda forma más frecuente de mastocitosis cutánea es la mastocitosis cutánea difusa. Afecta a todo el espesor de la piel, lo que produce un engrosamiento de la misma. Los más jóvenes con urticaria pigmentosa o con mastocitosis cutánea difusa pueden presentar erupciones y bullas con hemorragias asociadas (17).
El examen macroscópico de la piel puede mostrar lesiones típicas que en ocasiones son casi diagnósticas: signo de Darier, mastocitoma, eritema maculo-papular, eritrodermia difusa. Sin embargo, en cada caso, el diagnóstico se puede confirmar mediante una biopsia. La ausencia de lesiones cutáneas no confirma el diagnóstico de mastocitosis. En la enfermedad avanzada, puede detectarse una tendencia al sangrado (petequias espontáneas y/o hematomas), y son el resultado de la trombocitopenia y/o una alteración de la coagulación (por consumo o por hiperfibrinolisis).
El examen físico revela organomegalia en algunos pacientes con MS: linfadenopatía palpable, hepatomegalia y/o esplenomegalia. La infiltración de los órganos por los mastocitos puede conducir, no sólo a organomegalia, sino también a una función anómala de dicho órgano y el diagnóstico se puede confirmar mediante una biopsia.
Los hallazgos clínicos típicos (hallazgos-C) son: la malabsorción con pérdida de peso, hepatomegalia con ascitis, esplenomegalia con hiperesplenismo o fracturas patológicas secundarias a osteolisis (espontáneas). En la enfermedad severa (de alto grado), puede ocurrir anemia, trombocitopenia e infecciones recurrentes secundarias a insuficiencia medular (Tabla II). La ascitis es más frecuente en los casos de mastocitosis asociada a otra anomalía hematológica, y es de mal pronóstico y de difícil control. También puede producirse hipertensión portal, que se puede manejar mediante un shunt porta-cava. Cuando el hiperesplenismo conduce a una intensa pancitopenia, se recomienda hacer una esplenectomía.

La afectación ósea en la mastocitosis está relacionada con la infiltración de la médula ósea por los mastocitos, y está presente en el 70% de los casos. Radiológicamente, puede existir tanto lesiones líticas como blásticas, y, en ocasiones, coexisten ambas. La liberación de histamina por los mastocitos induce esclerosis, lo que explica la presencia de lesiones osteoblásticas, y la liberación de heparina y de prostaglandicnas induce lisis. Esto obliga a hacer el diagnóstico diferencial con otros procesos tales como la mielofibrosis, la enfermedad de Paget, las metástasis óseas o la fluorosis, en el caso de lesiones blásticas, o la osteoporosis quística, la talasemia o la enfermedad de Gaucher en las lesiones líticas (18). Las lesiones son difusas normalmente (85%), aunque también pueden ser focales (5%) o mixtas (10%). En la mayoría de los casos, las lesiones líticas son de pequeño tamaño, inferiores a 0,5 cm y no se acompañan de síntomas. Las lesiones difusas predominan en el esqueleto axial y típicamente combinan áreas de esclerosis con otras líticas (19). En unos pocos casos, sin embargo, se produce fracturas patológicas espontáneas secundarias a osteolisis extensa (20).
Estadificación complementaria
Dependiendo de la edad del paciente, los hallazgos clínicos y los parámetros de laboratorio, la estadificación de la mastocitosis debería incluir las siguientes pruebas: radiografía de tórax, serie ósea, ecografía abdominal y endoscopia digestiva, con toma de biopsia. La ecografía abdominal o la tomografía axial computerizada pueden revelar hepatomegalia, esplenomegalia o linfadenopatías, y sólo se solicitan en los casos en los que se sospeche una mastocitosis agresiva. La serie ósea puede mostrar la presencia de signos de osteoporosis, osteosclerosis o áreas de osteolisis focal. En la mayoría de los casos, las lesiones osteolíticas son pequeñas, y no se acompañan de síntomas clínicos. En otros casos, se puede producir fracturas patológicas espontáneas. Recientemente, Brumsen et al han publicado un artículo en que concluyen que el 9% de los casos de osteoporosis en hombres son secundarios a mastocitosis. El diagnóstico definitivo de muchas de las "osteoporosis idiopáticas" en varones podría, por tanto, conseguirse mediante un estudio anatomo-patológico de la médula ósea o incluso midiendo los niveles del metabolito N-metilhistamina en orina de 24 horas (21, 22).
Histología e inmunohistoquímitca
Para el estudio histológico de tejidos en los que se sospeche infiltración por mastocitos, se requiere de tinciones apropiadas, como la tinción de Giemsa, la tinción con azul de toluidina y tinciones con anti-triptasa. Junto a esto, los mastocitos pueden hacerse visibles empleando la reacción de la cloroacetato esterasa (CAE), enzima que no es exclusiva de los mastocitos (también puede detectarse en los neutrófilos) (23).
El diagnóstico de mastocitosis se basa tradicionalmente en la demostración de acúmulos focales de mastocitos con histología típica y determinadas propiedades citomorfológicas. Sin embargo, a veces resulta difícil diagnosticar la mastocitosis mediante la histología rutinaria.
La médula ósea es el órgano sistémico que se afecta con más frecuencia; el examen histológico de ésta puede demostrar datos típicos (Tabla III). Se recomienda el uso de la tinción de Giemsa y del azul de toluidina en los aspirados de médula ósea de los pacientes sospechosos. En algunos pacientes, el porcentaje de mastocitos en el aspirado de médula ósea es inferior al 5% e incluso, en ocasiones, inferior al 1%. Un porcentaje superior al 20% es casi diagnóstico de la leucemia de mastocitos.
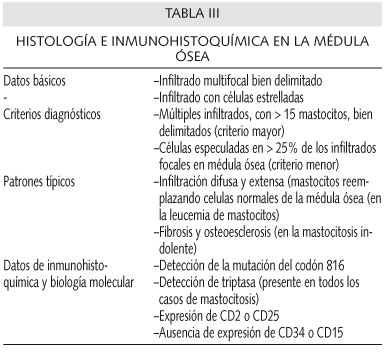
Al contrario de lo que ocurre en la médula ósea, estos criterios diagnósticos no se pueden aplicar a otros órganos. La presencia de algunos mastocitos en el bazo pueden ser suficientes para que se sospeche una mastocitosis, ya que normalmente no suele haber mastocitos en este órgano. Sin embargo, en otros órganos, como el tracto G-I, en los que la presencia de mastocitos es más frecuente, la detección de infiltrados densos de mastocitos no conduce directamente al diagnóstico de mastocitosis sistémica. Con indiferencia del órgano afecto, un infiltrado multifocal bien delimitado, o un infiltrado de células estrelladas, es diagnóstico de MS.
La triptasa y otros parámetros de laboratorio
Un marcador bien establecido y muy importante para el diagnóstico de mastocitosis sistémica son los niveles de triptasa. De hecho, los niveles séricos totales de triptasa reflejan la masa tumoral de mastocitos. En los pacientes sin afectación sistémica, los niveles son normales o están sólo levemente elevados; niveles elevados de triptasa aumentan la posibilidad de que se trate de una mastocitosis sistémica con afectación multiorgánica (la mayoría de los pacientes con mastocitosis sistémica tienen niveles de triptasa superiores a 20 ng/ml). En neoplasias mieloides, en especial, la leucemia mieloide crónica, también se han detectado niveles elevados de triptasa, e incluso pueden elevarse durante una reacción alérgica, por lo que no se trata de un marcador totalmente específico. Basándose en estas limitaciones, Valent et al (20), proponen como criterio diagnóstico de mastocitosis sistémica unos niveles de triptasa elevados de forma persistente, y Schwartz propone que para tener una alta sospecha de mastocitosis, no basta con que la triptasa sérica sea ≥ 20 ng/ml, sino que la relación entre ésta y la β-triptasa también sea ≥ 20, en ausencia de otros trastornos mieloides. En las anafilaxis, están elevadas tanto la triptasa sérica total (α y β), como la β-triptasa, pero la relación entre la total y la segunda es ≤ 10 (24). Aún se necesita más estudios para discernir si este criterio es más sensible o no que el hallazgo de mastocitos en médula ósea.
Aunque el nivel absoluto de triptasa sérica no predice la severidad de la enfermedad, puede proporcionar un método práctico para hacer un seguimiento de la masa de mastocitos tumorales a lo largo del tratamiento de la enfermedad. Además de la triptasa sérica, otros parámetros que pueden emplearse se basan en el hemograma, la bioquímica y la coagulación. En la enfermedad de bajo grado sin AHNMD, estas pruebas debieran ser totalmente normales, mientras que en la enfermedad agresiva o de alto grado, o en la AHNMD, pueden detectarse algunas anomalías, como citopenia, leucocitosis, eosinofilia, basofilia, monocitosis, LDH elevada, elevación de las enzimas hepáticas, hipoalbuminemia, alteración de los parámetros de la coagulación e incluso, mastocitos circulantes.
Mutaciones somáticas de C-Kit
Estudios con cultivos in vitro han demostrado que los mastocitos humanos derivan de células stem pluripotenciales CD34+ y que el factor de las células madre (SCF) es el factor de crecimiento fundamental en la proliferación, diferenciación y supervivencia de los mastocitos.
En la mayoría de los pacientes con MS de inicio en edad adulta se detecta una mutación de c-kit (mutación en el codón 816: Asp-816 → Val: D816V) (25). Por el contrario, la mastocitosis de la infancia no suele asociarse a dicha mutación, aunque se ha detectado en algunos casos pediátricos aislados, al igual que la mutación c-kit Gly-839 → Lys. Esto indica que la MS del adulto y la pediátrica son dos entidades totalmente distintas (26).
Curiosamente, en los pacientes con MS, la mutación c-kit no sólo se detecta en los mastocitos, sino que también puede detectarse en otras líneas celulares, incluyendo los monocitos o incluso las células B. En los casos con mieloproliferación asociada (estado asintomático), la mutación está presente incluso en las células mononucleares de la sangre periférica. En un subgrupo de pacientes con una variante "smoldering" de la mastocitosis, la mutación no sólo se detecta en los mastocitos, sin también en las células que no pertenecen a la línea de las células hematopoéticas de los mastocitos (10).
El análisis de la heterogeneidad de esta enfermedad y ejemplos específicos de cómo progresan, sugieren que debe haber al menos 4 posibilidades que expliquen la diversidad de las manifestaciones clínicas en pacientes con mastocitosis. La primera de estas posibilidades es que una mutación activadora en el codón 816 sea efectiva para promover la división exagerada de mastocitos en algunos individuos, en parte, debido al polimorfismo genético del huésped. Otra posibilidad es que una mutación activante en la posición 816 actúe como una "mutación permisiva" que permita que la enfermedad se desarrolle si tiene lugar una segunda mutación (o incluso una tercera) en vías críticas. En algunos pacientes, puede haber una segunda mutación independiente de la primavera en otro grupo de células, originando un segundo clon maligno. La última posibilidad analizada es que exista inestabilidad cromosómica que predisponga al paciente tanto a la mutación 816 como a otros eventos (27).
Diagnóstico y criterios definitorios
Se ha formulado una serie de criterios mayores y menores, basados en hallazgos clínicos, histológicos, inmunohistoquímicos y de laboratorio, con el fin de diagnosticar una mastocitosis sistémica y de discriminar entre este diagnóstico y el de mastocitosis cutánea, mastocitoma solitario, sarcoma de mastocitos, hiperplasia de mastocitos sistémica y aumento de mastocitos en una neoplasia mielomatosa sin mastocitosis (Fig. 2). El diagnóstico de mastocitosis sistémica se consigue cuando se cumple un criterio mayor y uno menor, o si coexisten los 3 criterios menores. Una vez llegado al diagnóstico, el porcentaje de infiltración de mastocitos en la médula ósea permitirá saber si se trata o no de una leucemia de mastocitos (> = 20% de infiltración). Cuando la infiltración sea inferior al 20%, si no existe hallazgos B ni C, se hablará de mastocitosis indolente; si sólo hay hallazgos B, mastocitosis sistémica, y cuando sólo haya hallazgos C, mastocitosis sistémica agresiva.

En los niños con mastocitosis, el principal hallazgo es la lesión cutánea, que suele desaparecer a lo largo de la adolescencia; por este motivo, si el niño no tiene organomegalia ni una alteración importante en sangre periférica, no será necesario tomar una biopsia de médula ósea. (20). Sin embargo en adultos, incluidos aquellos en los que la mastocitosis se haya iniciado antes de la pubertad, el estudio diagnóstico debe incluir una biopsia de médula ósea (28).
Tratamiento de la mastocitosis sistémica
En la actualidad, no existe tratamiento curativo de la mastocitosis. Los pacientes con mastocitosis indolente suelen tener una progresión lenta a lo largo del tiempo. Según las actuales recomendaciones, estos pacientes deben ser monitorizados de forma estrecha y cuidadosa, pero no se deben tratar con citostáticos.
En los pacientes con mastocitosis sistémica y lesiones óseas significativas, una opción razonable consiste en el tratamiento combinado de IFN-alfa-2b (subcutáneo) y prednisolona (oral) (29). Normalmente se inicia el tratamiento con prednisolona (50-75 mg por día) durante algunos días, y después, se añade IFN-alfa2b adicional comenzando con una dosis de 3 millones de unidades internacionales (UI) tres veces a la semana. Durante los primeros días de tratamiento con IFN, los pacientes deben ser monitorizados en el hospital. Después de unas pocas semanas, la dosis de IFN puede aumentarse 3-5 millones de unidades por día y la prednisolona, reducirse hasta alcanzar una dosis de mantenimiento de 12,5 mg/día o inferior, incluso hasta suspenderse. En los pacientes con una osteoporosis severa y fracturas patológicas, parece preferible la administración de IFN-alfa-2b sin glucocorticoides, dados los efectos secundarios de los mismos. En los pacientes que respondan, el tratamiento debería mantenerse hasta que haya signos o síntomas de toxicidad o efectos secundarios o que exista evidencia de progresión. En una revisión de la literatura disponible, Valent y cols. (30) describieron 14 pacientes con mastocitosis agresiva tratados con IFN-alfa con o sin prednisolona. De estos, el 21,4% (3 pacientes) presentaron una respuesta mayor con desaparición de los hallazgos-C. En cinco pacientes (35,7%) se obtuvo una respuesta parcial, y el 42,9% de los pacientes no respondió. Las tasas de respuestas fueron similares en los que recibieron la combinación con corticoides que en los que no los recibieron. Se ha descrito algún caso de respuestas casi completas en pacientes tratados con IFN-alfa a altas dosis durante largos periodos de tiempo, con una tolerancia aceptable al mismo (31).
Cuando el IFN no es suficiente para el control de la enfermedad, se emplea una amplia variedad de citostáticos (cladribina, arabinósido de citosina, daunorrubicina o hidroxiurea) (32).
Si la situación lo permite y se produce una respuesta mayor a la quimioterapia de inducción, estos pacientes son subsidiarios de un transplante de médula ósea. Si no se puede aplicar el transplante de médula ósea, los pacientes en remisión deberían recibir tratamiento de consolidación.
Durante el tratamiento con quimioterapia, la monitorización se hará basándose en una serie de parámetros: los relacionados con los hallazgos-C (elevación de enzimas hepáticas, la elevación del calcio en sangre o el descenso de la hemoglobina) y los que reflejan la carga tumoral de los mastocitos neoplásicos (la histología de la médula ósea, los niveles de triptasa, CD25). En función a estos datos, se habla de respuesta clínica pura cuando sólo se produzca mejoría en los parámetros relacionados con los hallazgos C; de respuesta completa, cuando se normalicen ambos grupos de parámetros; y de ausencia de respuesta cuando ocurra lo contrario (30).
En la actualidad se está empleando algunos agentes derivados de la biología molecular. Así, por ejemplo, se ha empleado fármacos tales como el Imatinib (STI571), que ha demostrado ser útil en los casos en los que exista determinadas mutaciones, como la c-kit Phe522Cys, pero no útil en otras como la Asp816Val, que es la más frecuente en estos tumores (33).
Fármacos complementarios al tratamiento quimioterápico
A todos los pacientes con mastocitosis agresiva se les administra antagonistas anti-H1 y anti-H2, y/o prednisolona previo al inicio del tratamiento con los agentes quimioterápicos, ya que los mediadores de los mastocitos pueden causar síntomas significativos (34). Estos pacientes se monitorizan cuidadosamente en el hospital durante los primeros días de tratamiento. En los pacientes con síntomas severos y recurrentes asociados a los mediadores, con riesgo de desarrollo de un shock cardiovascular, se recomienda que se disponga de epinefrina para su autoadministración por si fuera necesario.
El tratamiento de la enfermedad intestinal es variable. Los anticolinérgicos pueden proporcionar cierto alivio. La cromolina sódica oral puede ser útil en el manejo de los síntomas abdominales. Los glucocorticoides sistémicos son efectivos en pacientes con malabsorción severa. También la ascitis exudativa ha sido manejada con éxito mediante glucocorticoides sistémicos.
Leucemia de mastocitos
La leucemia de mastocitos es una rara enfermedad caracterizada por una rápida expansión de mastocitos en la médula ósea, la sangre y en otros órganos, con la consiguiente organopatía (35). El pronóstico es grave, y la supervivencia, mínima. No hay ningún tratamiento efectivo para estos pacientes. Incluso la quimioterapia convencional no produce una respuesta duradera en estos pacientes, por lo que se está empleando regímenes agresivos, como los utilizados en la leucemia mieloide aguda. Si existe un donante disponible y se ha obtenido una remisión después de la quimioterapia, también son candidatos a transplante de médula ósea.
Neoplasias malignas hematológicas asociadas
Aproximadamente en un 15-30% de los pacientes con MS se observa una neoplasia hematológica asociada, como síndromes mieloproliferativos, síndromes mielodisplásicos o leucemia mieloide aguda. A veces resulta difícil distinguir entre la neoplasia hematológica asociada y la mastocitosis. La opción terapéutica más recomendada consiste en planificar el tratamiento de la neoplasia maligna hematológica como si no existiera MS asociada, y el tratamiento de la MS como si no existiera la otra patología, y posteriormente, adaptar el tratamiento; en ocasiones, el tratamiento para ambos tratamientos resulta ser el mismo.
Conclusiones
La mastocitosis sistémica es un proceso más frecuente de lo que realmente detectamos en la clínica. Incluso en pacientes con una mastocitosis típica, la mediana de tiempo desde el inicio de los síntomas hasta el diagnóstico es de 1 año. Se debe sospechar ante cualquier paciente con síncopes repetitivos, en especial si no existe otra causa que lo justifique, en varones con osteoporosis y en pacientes con lesiones óseas osteblásticas y/u osteolíticas de etiología no filiada. En estos casos, el primer examen a realizar es la determinación de niveles sanguíneos de triptasa, que, por sí solos, no serían diagnósticos de esta enfermedad. Si dichos niveles permanecieran elevados pasadas algunas semanas, posteriormente es mandatorio hacer un estudio de médula ósea. En ocasiones, un diagnóstico precoz puede evitar situaciones que desencadenen posteriormente un shock a los pacientes afectos. El estudio de médula ósea, junto con una alta sospecha clínica, son la base para el diagnóstico precoz y definitivo.
Bibliografía
1. Metcalfe DD. Classification and diagnosis of mastocitosis: Current status. J Invest Dermatol 1991; 96: 2S-4S. [ Links ]
2. Lawrence JB, Friedman BS, Travis WD, Chinchilli VM, Metcalfe DD, Gralnick HR. Hematologic manifestations of systemic mast cell disease: a prospective study of laboratory and morphologic features and their relation to prognosis. Am J Med 1991; 91: 612-24. [ Links ]
3. Valent P, Sperr WR, Schwartz LB, Horny HP. Diagnosis and classification of mast cell proliferative diroders: Delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol 2004; 114: 3-11. [ Links ]
4. Mitsui H, Furitsu T, Dvorak AM, Irani AA, Schwartz LB, Inagaki N, et al. Development of human mast cells from umbilical cord blood cells by recombinant human and murine c-kit ligand. Proc Natl Acad Sci USA 1993; 90: 735. [ Links ]
5. Valent P. The riddle of the mast cell: c-kit ligand and missing link? Immunol Today 1994; 15: 111-4. [ Links ]
6. Rottem M, Okada T, Goff JP, Metcalfe DD. Mast cells cultured from peripheral blood of normal donors and patients with mastocytosis originate from a CD34+/FceRI- cell population. Blood 1994; 84: 2489-96. [ Links ]
7. Sillaber C, Strobl H, Bevec D, Ashman LK, Butterfield JH, Lechner K et al. IL-4 regulates c-kit proto-oncogene product expression in human mast and myeloid progenitor cells. J Immunol 1991; 147: 4224-8. [ Links ]
8. Feger F, Ribadeau Dumas A, Leriche L, Valent P, Arock M. Kit and c-kit mutations in mastocytosis: a short overview with special reference to novel molecular and diagnostic concepts. Int Arch Allergy Immunol 2002; 127: 110-4. [ Links ]
9. Akin C, Kirschenbaum AS, Semere T, Worobec AS, Scout LM, Metcalfe DD. Analysis of the surface expression of c-kit and occurrence of the c-kit Asp816Val activating mutation in T cells, B cells, and myelomonocytic cells in patients with mastocytosis. Exp Hematol 2000; 28: 140-7. [ Links ]
10. Yavuz AS, Lipsky PE, Yavuz S, Metcalfe DD, Akin C. Evidence for the involvement of a hematopoietic progenitor cell in systemic mastocytosis from single cell analysis of mutations in the c-kit gene. Blood 2002; 100: 661-5. [ Links ]
11. Shimizu Y, Sakai K, Miura T, Narita T, Tsukagoshi H, Satoh Y et al. Characterization of adult-type mast cells derived from human bone marrow CD34+ cells cultured in the presence of stem cell factor and interleukin-6. Interleukin-4 is not required for constitutive expression of CD54, FceRIa and chymase, and CD13 expression is reduced during differentiation. Clin Exp Allergy 2002; 32: 872-80. [ Links ]
12. Sperr W, Jordan JH, Fiegl M, Escribano L, Dirnhofer S, Semper H et al. Serum tryptase levels in patients with mastocytosis correlation with mast cell burden and implication for defining the category of disease. Int Arch Allergy Immunol 2002; 128: 133-6. [ Links ]
13. Kepley CL, Pfeiffer JR, Schwartz LB, Wilson BS, Oliver JM. The identification and characterization of umbilical cord blood-derived human basophils. J Leukocyte Biol 1998;64:474. [ Links ]
14. Kepley CL, Craigg SS, Schwartz LB. Identification and partial characterization of a unique marker for human basophils. J Immunol 1995; 154: 6548. [ Links ]
15. McEuen AR, Buckley MG, Compton SJ, Walls AF. Development and characterization of a monoclonal antibody specific for human basophils and the identification of a unique secretory product of basophil activation. Lab Invest 1999; 79: 27. [ Links ]
16. Li LX, Li Y, Reddel SW, Cherrian M, Fiend DS, Stevens RL et al. Identification of basophilic cells that Express mast cell granule proteases in the peripheral blood of asthma, allergy, and drug-reactive patients. J Immunol 1998; 161: 5079. [ Links ]
17. Soler NA. Mastocytosis and the skin. Hematol Oncol Clin North Am 2000; 14: 557. [ Links ]
18. Siegel S, Sadler MA, Yook C, Chang V, Miller J. Systemic mastocitosis with involvement of the pelvis: A radiographic and clinicopathologic study-a case report. Clin Imaging 1999; 23: 245-8. [ Links ]
19. Inaoui R, Petit B, Jaccard A, Bertin P, Trèves R. Aggressive systemic mastocitosis. Joint Bone Spine 2003; 70: 64-6. [ Links ]
20. Valent P, Horny H-P, Escribano L, Longley BJ, Li CY, Scwartz LB, et al. Diagnostic criteria and classification of mastocitosis: A consensus proposal. Leukemia Res 2001; 25: 603-25. [ Links ]
21. Brumsen C, Papapoulos SE, EGWM Lentjes, PM Kluin and NAT Hamdy. A potencial role for the mast cell in the pathogenesis of idiopathic osteoporosis in men. Bone 2002; 31: 556-61. [ Links ]
22. Pusl T, Kenngott S, Bartl R, Baur A, Ludolph-Hauser D, Juengst D. A case of systemic mastocitosis associated with severe osteoporosis and pathologic fractures. Eur J Int Med 2004; 15: 537-9. [ Links ]
23. Li CY. Diagnosis of mastocitosis: Value of cytochemistry and inmunochemistry. Leuk Res 2001; 25: 603-25. [ Links ]
24. Schwartz LB. Clinical utility of tryptase levels in systemic mastocytosis and associated hematologic disorders. Leuk Res 2001; 25: 553-62. [ Links ]
25. Longley BJ, Metcalfe DD. A proposed classification of mastocitosis incorporating molecular genetics. Hematol Oncol Clin North Am 2000; 14: 697-701. [ Links ]
26. Longley BJ, Metcalfe DD, Tharp M, Wang X, Tyrrell L, Lu SZ, et al. Activating and dominant inactivating c-kyt catalytic domain mutations in distinct forms of human mastocitosis. Proc Natl Acad Sci USA 1999; 96: 1609-14. [ Links ]
27. Metcalfe DD, Akin C. Mastocytosis: molecular mechanisms and clinical disease heterogeneity. Leuk Res 2001; 25: 577-82. [ Links ]
28. Horny HP, Valent P. Histopathological and immunophenotypical aspects of mastocytosis. Int Arch Allergy Immunol 2002; 127: 115-7. [ Links ]
29. Worobec AS, Metcalfe DD. Mastocitosis: Current treatment concepts. Int Arch Allergy Immunol 2002; 127: 153-5. [ Links ]
30. Valent P, Akin C, Sperr WR, Horny HP, Metcalfe DD. Aggressive systemic mastocitosis and related mast cell disorders. Current treatment options and proposed response criteria. Leukemia Res 2003; 27: 635-41. [ Links ]
31. Butterfield JH, Tefferi A, Kozuh GF. Successful treatment of systemic mastocitosis with high-dose interferon-alfa: Long-term follow-up of a case. Leuk Res 2005; 29: 131-4. [ Links ]
32. Escribano L, Pérez de Oteyza J, Núñez R, Orfao A. Cladribina induces immunophenotypical changes in bone marrow mast cells from mastocitosis. Report of a case of mastocitosis associated with a lymphoplasmocytic lymphoma. Leuk Res 2002; 26: 1043-6. [ Links ]
33. Akin C, Brockow K, D'Ambrosio C, Kirshenbaum AS, Ma Y, Longley BJ, et al. Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated c-kit. Exp Hematol 2003; 31: 686-92. [ Links ]
34. Worobec AS. Treatment of systemic mast cell disorders. Hematol Oncol Clin North Am 2000; 14: 659. [ Links ]
35. Sperr WR, Horny HP, Lechner K, Valent P. Clinical and biologic diversity of leukemias occurring in patients with mastocitosis. Leuk Lymphoma 2000; 37: 473-86. [ Links ]
36. Valent P, Akin C, Sperr WR, Horny HP, Arock M, Lechner et al. Diagnosis and treatment of systemic mastocytosis: State of the art. Br J Haematol 2003; 122: 1-23. [ Links ]
37. Castells M, Austen KF. Mastocytosis: Mediator-related signs and symptoms. Int Arch Allergy Immunol 2002; 127: 147-152. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
María José Molina Garrido.
Hospital General Universitario de Elche.
Camino de la Almazara, 11.
03203 Elche, Alicante.
e-mail: mjmolinagarrido@hotmail.com
Trabajo aceptado: 11 de octubre de 2007

