










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkThe European Journal of Psychiatry
versión impresa ISSN 0213-6163
Eur. J. Psychiat. vol.27 no.3 Zaragoza jul./sep. 2013
https://dx.doi.org/10.4321/S0213-61632013000300005
Safety and Tolerability of Atomoxetine Hydrochloride in a Long-Term, Placebo-Controlled Randomized Withdrawal Study in European and Non-European Adults with Attention-Deficit/ Hyperactivity Disorder
Angelo Camporeale, MD*; Himanshu Upadhyaya, MBBS, MS*; J. Antoni Ramos-Quiroga, MD, PhD**,***; David Williams, MS****; Yoko Tanaka, PhD*; Jeannine R. Lane, PhD****; Rodrigo Escobar, MD*; Paula Trzepacz, MD* and Albert J Allen, MD, PhD*
*Lilly Research Laboratories, Lilly Corporate Center, Indianapolis, USA
**Department of Psychiatry, Hospital Universitari Vall d'Hebron, CIBERSAM Barcelona, Spain
***Department of Psychiatry and Legal Medicine, Universitat Autònoma de Barcelona, Bellaterra, Spain
****inVentiv Health Clinical, Indianapolis, USA
ABSTRACT
Background and Objectives: Safety and tolerability of atomoxetine were studied in the largest double-blind, placebo-controlled, randomised withdrawal trial of atomoxetine (80 or 100 mg/day) in adults with attention-deficit/hyperactivity disorder (ADHD).
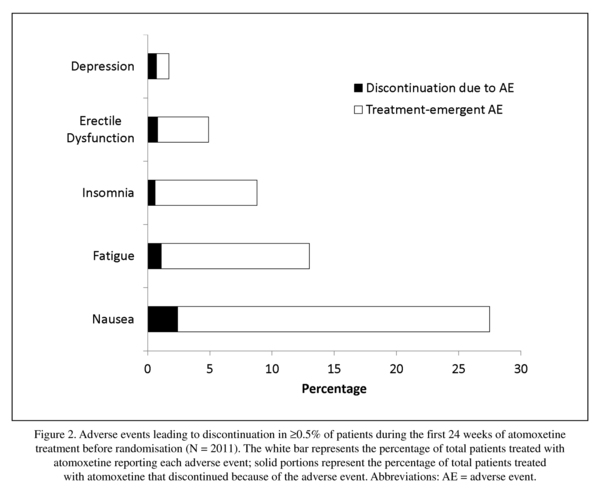
Methods: Patients (N = 2017), 18 to 50 years of age, with ADHD were enrolled from 18 countries. Patients who responded to atomoxetine during a 12-week open-label treatment phase and maintained that response during a 12-week double-blind maintenance phase were randomised to atomoxetine (N = 266) or placebo (N = 258) for 25 weeks of double-blind treatment. Treatment differences were compared for serious adverse events (AEs), treatment-emergent AEs (TEAEs), discontinuation due to AEs, vital signs, body weight, and electrocardiograms.
Results: During the 25-week double-blind treatment phase, discontinuations due to AEs were similar between atomoxetine and placebo (3.4% vs. 1.9%; P = .418). The percentage of patients experiencing >1 TEAE(s) was significantly higher for atomoxetine than placebo (47.0% vs. 37.6%; P = .034), but there were no significant differences for any individual TEAE. Diastolic blood pressure (-0.1 vs. -2.3 mmHg), heart rate (-1.4 vs. -5.3 bpm), and weight (-0.2 vs. 1.1 kg) were significantly different between atomoxetine and placebo (P <.001). There were no significant differences between atomoxetine and placebo in the frequencies of patients showing an increase from baseline >30 ms in Fridericia's QT correction (QTcF; 1.4% vs. 2.6%) or Bazett's QT correction (QTcB; 2.8% vs. 2.6%). During the entire study, no patient had a QTcF or QTcB >500 ms, or an increase from baseline >60 ms.
Conclusions: This study demonstrated that atomoxetine exhibited an acceptable safety profile in adults with ADHD after 1 year of treatment, and no clinically meaningful safety-related rebound effects were observed following abrupt discontinuation after 24 weeks of treatment.
Key words: Attention-deficit/hyperactivity disorder; Adult; European; Atomoxetine.
Introduction
There have been major advances in the treatment and understanding of attention-deficit/hyperactivity (ADHD) during the last decade1-6. Yet despite this growing understanding, ADHD in adults is currently underdiagnosed in many European countries, leading to ineffective treatment and higher costs of illness7.
Of the multiple pharmacotherapeutic options for treatment of ADHD in adults8-15, the stimulants methylphenidate and dexamphetamine are the most widely used in Europe7; however, they are only approved in some countries. Although efficacy and safety of these stimulants are well-established, as many as 30% of patients have inadequate response, tolerability problems (e.g., sleep disturbance) or contraindicated comorbidities (e.g., schizophrenia, hyperthyroidism, cardiac arrhythmias, angina pectoris, and glaucoma)16,17. Additionally, even with extended-release formulations, the stimulants remain limited in their duration of effect, and the abuse liability may lead to reluctance to use them16,17. Hence, treatment should be selected according to patient need for lasting efficacy, comorbidity profile, and risk of abuse of prescription medications.
Atomoxetine hydrochloride, a selective norepinephrine reuptake inhibitor, is a nonstimulant medication approved to treat ADHD in adults in North America and several other countries17. Because of its demonstrated efficacy, limited spectrum of adverse effects, and absence of abuse potential, atomoxetine may aid in treating patients with ADHD. Indeed, multiple studies in children18-23 and adults8,10,11,24,25 have shown that atomoxetine has an acceptable safety profile, with most adverse events (AEs) having low-to-moderate severity, which diminish over the first month of treatment. To extend these findings, the long-term safety and efficacy of atomoxetine in adults with ADHD were examined in a double-blind, placebo-controlled randomized withdrawal study. Patients who responded to atomoxetine during a 12-week open-label treatment phase and maintained that response during a 12-week double-blind maintenance phase were randomized to an additional 25 weeks of atomoxetine or placebo during a double-blind randomized withdrawal phase. The efficacy results from this trial, reported in Upadhyaya et al.26, demonstrated substantial treatment responses to atomoxetine during the 12 week open-label and 12-week double-blind maintenance periods as measured by a ≥30% continuous reduction from baseline in Conners' Adult Attention-Deficit/Hyperactivity Disorder Rating Scale (CAARS)-Investigator Screening Version (CAARS-Inv:SV)27 total score and a rating ≤3 (minimally ill) on the Clinical Global Impression ADHD-Severity (CGI-ADHD-S)28. In the 25-week randomized withdrawal phase that followed, these authors showed that atomoxetine was able to effectively control ADHD symptoms as demonstrated by a significantly greater proportion of atomoxetine- than placebo-treated patients maintaining their treatment responses. In this paper, we report the 24-week prerandomisation and 25-week postrandomisation safety results from this trial.
Method
Participants
This study was conducted at 152 centers in 18 countries throughout Asia, North America, South America, and Europe. Eligible patients were age 18 to 50 years and met the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition Text RevisionTM (DSM-IV-TR) criteria for current and childhood ADHD as assessed by the Conners' Adult ADHD Diagnostic Interview for DSM-IV27; had a score of ≥2 on at least 6 items of either the inattentive or hyperactive core subscales of the CAARS-Inv:SV27 and of the CAARS-Observer Screening Version (CAARS -O:SV) with adult ADHD prompts for current symptoms27; and had a score of ≥20 on CAARS-Inv:SV 18-item total ADHD symptom score. Patients were also required to have a CGI-ADHD-S28 rating of ≥4 (moderately ill) at Visits 1 and 2. The CGI-ADHD-S was rated on a 7-point scale with 1 meaning "normal, not at all ill" to 7 meaning "among the most extremely ill patients".
Patients who met DSM-IV-TR diagnostic criteria for any history of bipolar disorder, current major depression, a current anxiety disorder (including generalised anxiety disorder, panic disorder, or social phobia), or any history of a psychotic disorder were excluded from the study. Patients who were currently using alcohol, drugs of abuse, or any prescribed or over-the-counter medication in a manner that the investigator considered indicative of chronic abuse or who met DSM-IV-TR criteria for alcohol or other substance dependence were also excluded. Additional details about the inclusion and exclusion criteria are provided in Upadhyaya et al26.
An informed consent document approved by an Ethical Review Board or similar body was signed by the patient and/or representative and deemed appropriate, according to local laws and regulations. The study was conducted in accordance with the Declaration of Helsinki and the applicable laws and regulations of the study countries and regions.
Study Design
In this study, a randomised, double-blind, maintenance of response (randomised withdrawal) design was implemented to test the hypothesis that atomoxetine (80 or 100 mg/ day) was superior to placebo in maintaining a satisfactory response at the end of double-blind treatment in adult outpatients with ADHD. The study design was made up of 3 study periods and is illustrated in Figure 1.
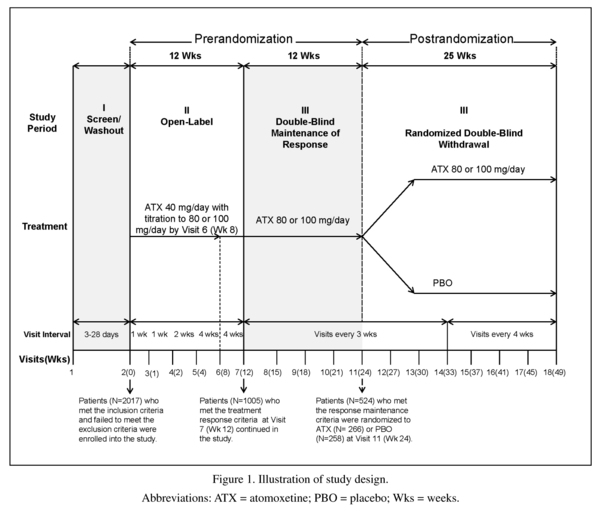
The first study period was a screening/ washout phase. The second study period was a 12-week open-label atomoxetine-treatment phase (Visits 2 to 7 [Weeks 0 to 12]). During this study period, patients received open-label atomoxetine at a starting dose of 40 mg/day (Visit 2 [Week 0]) after completion of all assessments, with titration to a target dose of 80 or 100 mg/day (depending on tolerability) by Visit 6 (Week 8). This dose was maintained up to Visit 7 (Week 12). At Week 12, patients were classified as responders if they had a ≥30% reduction in their baseline (Visit 2) CAARS-Inv:SV total score, and a CGI-ADHD-S score ≤3 (minimally ill). These criteria are commonly used in ADHD studies and provide a clinically meaningful level of improvement24,29.
The third study period began with a 12-week (Visits 7 to 11 [Weeks 12 to 24]) double-blind maintenance phase with atomoxetine (80 or 100 mg/day). Only atomoxetine responders were allowed to enter this study period; non-responders were discontinued from the study. During this study period, atomoxetine responders were required to continuously maintain their initial treatment responses for 3 months after assessment at Visit 7 (Week 12) and through assessment at Visit 11 (Week 24), except for 1 excursion. An "excursion" was a brief shift in the patient's symptoms or behavior that did not affect the core ADHD symptoms, but was reflected in the patient's overall CGI-ADHD-S score. An excursion was defined as a change (worsening) in the CGI-ADHD-S ≤1 point from the patient's Visit 7 score, not to exceed a score of 4 (moderately ill) and a CAARS-Inv:SV total ADHD symptom score that still met the response criterion (reduction ≥30% in the baseline [Visit 2] score).
The last portion of the third study period was a 25-week double-blind, randomized withdrawal phase in which atomoxetine responders who were able to maintain their treatment responses were randomized in a 1:1 ratio to atomoxetine (80 or 100 mg/day) or placebo. During this study period, patients must have continuously met the response criteria for 25 weeks, after assessment at Visit 11 (Week 24) and through assessment at Visit 18 (Week 49), except for a total of 2 excursions. Patients were allowed 1 excursion after assessment at Week 24 and through assessment at Week 37 (13 weeks) and another excursion after assessment at Week 37 and through assessment at Week 49 (12 weeks). Patients were not permitted to have excursions at 2 consecutive visits. To be considered a relapser, patients must have had 2 consecutive visits with a CGI-ADHD-S score ≥4 points and a return to ≥80% of the patient's baseline (Visit 2) CAARS-Inv:SV total score after Visit 11 (Week 24). If, at any single visit, a patient showed evidence of a return of symptoms that met the severity criteria and, because of worsening symptoms, was unwilling to remain in the study or did not return for a second visit, the patient was also considered to have relapsed and was discontinued from the study.
Safety Assessments
Safety measures recorded at every visit included spontaneously reported AEs, weight, and sitting blood pressure (BP) and heart rate (HR). Treatment-emergent AEs (TEAEs) were defined as events that first occurred or worsened after study entry, as compared with the maximum severity at or before randomisation. Serious AEs (SAEs) were defined as any AE that met one of the following criteria: death; initial or prolonged inpatient hospitalisation; a life-threatening experience (i.e., immediate risk of dying); persistent or significant disability/incapacitation; congenital anomaly/birth defect; or an event considered significant by the investigator for any other reason. All SAEs and TEAEs were reported according to the Medical Dictionary for Regulatory Activities (MedDRA), Version 14.
Sitting vital signs were measured at every visit. Investigators at each clinical trial site were instructed to measure sitting BP twice at each visit with at least 5 minutes in between each measurement, and report the mean of the 2 measurements. Treatment-emergent abnormally high systolic BP (SBP) was defined as an increase ≥20 mmHg from baseline to a value ≥160 mmHg, and abnormally low was defined as a decrease ≥20 mmHg from baseline to a value ≤90 mmHg. Abnormally high diastolic BP (DBP) was defined as an increase ≥10 mmHg from baseline to a value ≥100 mmHg, and abnormally low was defined as a decrease ≥10 mmHg from baseline to a value ≤50 mmHg. Abnormally high HR was defined as an increase ≥15 bpm from baseline to a value >120 bpm, and abnormally low was defined as a decrease ≥15 bpm from baseline to a value <50 bpm.
Weight was measured at baseline (screening), Visit 7, Visit 15, and the final study visit. Treatment-emergent abnormal weight gain was defined as ≥7% increase from baseline. Abnormal weight loss was defined as ≥7% decrease from baseline. An electrocardiogram (ECG) was collected at baseline (Visit 1 or between Visit 1 and Visit 2), Visit 7, and the final study visit (including early termination). Standard laboratory tests, including chemistry, hematology, and urinalysis panels, were collected at baseline. Other laboratory tests such as a urine drug screen, thyroid function test, and pregnancy tests (urine and serum), were completed at baseline. Follow-up laboratory measurements were not planned, and only conducted at the discretion of the local investigator. Therefore, analyses of laboratory data are not presented.
Prior to administration of study drug at Visit 2 and at each subsequent visit, suicidality was evaluated by a study physician using the Columbia Suicide-Severity Rating Scale (C-SSRS)30. Additional safety measures included the Hamilton Anxiety Rating Scale-14 items (HAMA)31,32 and the Hamilton Depression Rating Scale-17 items (HAMD-17)33,34, which were collected at baseline, Visit 7, and the final study visit.
Statistical Analyses
Safety analyses were performed on the safety analysis set, which included data from all patients receiving at least 1 dose of the study drug according to the treatment the patients actually received. Safety data were analyzed prerandomisation (open-label treatment period combined with the double-blind maintenance period [i.e., 24 weeks atomoxetine]) and postrandomisation (double-blind randomised withdrawal period [i.e., 25 weeks atomoxetine or placebo]). For the 24-week prerandomisation analyses, baseline was considered the last available observation of Visits 1 and 2. For the 25-week, double-blind postrandomisation analyses, baseline was considered the last measurement obtained on or before the randomisation visit. Exposure to study drug was summarized prerandomisation and postrandomisation.
Categorical variables were summarized. Postrandomisation treatment differences were compared with Fisher's exact test. For predefined abnormalities, only patients with a normal baseline value were included in the analysis. Post hoc analyses of the incidence and persistence of the most common AEs were performed postrandomisation.
Wilcoxon signed-ranked tests were used to assess within-group changes from baseline for continuous variables. Postrandomisation treatment differences for these measures were analyzed using an analysis of covariance model with the terms treatment, investigator, and baseline; however, the HAMA and HAMD-17 were analyzed using an analysis of variance model with the terms for treatment and investigator. For early dropout patients, endpoint was imputed using the last-observation-carried-forward approach. Changes from baseline in QT corrections were performed using the Bazett's (QTcB) and Fridericia's (QTcF) QT corrections.
All tests were 2-sided and conducted at the 0.05 alpha level. All statistical analyses were performed using SAS, version 9.1 (SAS Institute, Inc, Cary, NC). These analyses were performed on an intent-to-treat basis.
Results
Patient Disposition, Demographics, and Discontinuation
Of the 2534 patients screened, 2017 were enrolled into the study and 517 failed screening. Of the 2017 enrolled patients, 1493 discontinued prerandomisation (open-label atomoxetine and double-blind maintenance periods combined) for reasons such as AEs (17.3%), response maintenance criteria not met (16.0%), subject decision (12.1%), protocol violation (11.4%), and lost to follow-up (7.4%), lack of efficacy (6.0%), critical relapse (2.8 %), physician decision (0.5 %), missing (0.2 %), sponsor criteria not met (0.1 %), and entry criteria not met (0.1 %). In total, 524 patients, who met the response maintenance criteria at Visit 11 were randomized to atomoxetine (N = 266) or placebo (N = 258). Of these, 184 (69.2%) atomoxetine-treated and 165 (64.0%) placebo-treated patients completed the study.
Prerandomisation, 2011 patients received ≥1 dose of atomoxetine (mean exposure = 105.2 days). Mean exposure to atomoxetine postrandomisation was 158.9 days. Most patients were White and male (Table 1; see Upadhyaya et al.26 for additional patient characteristics). Postrandomisation, patient demographics were similar between the treatments (Table 1).
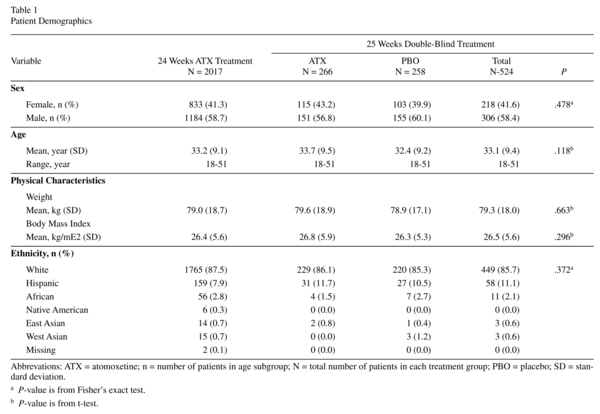
Figure 2 shows AEs leading to discontinuation reported with ≥0.5% frequency prerandomisation. Postrandomisation reasons for discontinuation were similar between atomoxetine and placebo with the exceptions of lack of efficacy (1.1% vs. 4.3%; P= .031) and response maintenance criteria not being met (0.0% vs. 1.9%; P= .028). Overall incidence of discontinuations due to an AE was similar between atomoxetine and placebo (3.4% vs. 1.9% P= .418), and no single AE was predominant (depression and myocardial infarction were 0.4% vs. 0.4%; depressed mood, ejaculation disorder, erectile dysfunction, hyperhidrosis, hypertension, nausea, and oral disorder were 0.4% vs. 0.0%; breast disorder, tremor, and weight increased were 0.0% vs. 0.4%).
Serious Adverse Events
Twenty-nine of 2011 (1.4%) patients experienced 35 SAEs prerandomisation. Six6 patients each experienced 2 SAEs and 23 patients each experienced a single SAE. The most common SAE was accidental overdose of study drug (0.2%). Of the 35 SAEs, 8 were judged by the investigator to be treatment-related (alcohol abuse and 2 events of restlessness in 1 patient; haemorrhage and headache in 1 patient; bradykinesia, suicidal ideation; palpitations; and auditory hallucination). All 5 patients who experienced these SAEs discontinued from the study.
Of the 524 randomized patients, there was no significant difference in the incidence of SAEs between atomoxetine and placebo (2.6% vs. 1.6%; P= .55). Seven patients receiving atomoxetine each experienced a single SAE and 4 patients receiving placebo each experienced a single SAE. Of the 11 SAEs, 1 (myocardial infarction) experienced by a placebo-treated patient was judged by the investigator to be related to study treatment; the patient discontinued from the study.
One death (reported as suspected myocardial infarction by the principal investigator at the study site) occurred at Visit 14 in a male patient. There is no confirmation of the diagnosis; it is unknown at the time of this report if an autopsy was conducted. The patient received open-label atomoxetine at Visits 2 and 3 (40 mg/day), Visit 4 (80 mg/day), and from Visits 5 to 6 (100 mg/day). From Visits 7 to 14, the patient continued to receive blinded atomoxetine (100 mg/day). The investigator was unable to assess the relatedness between this event and blinded study drug, open-label atomoxetine, or protocol procedures. The patient had a partial block of the right bundle branch (heart) as seen by the investigator in the ECG at the time of enrollment and in a second ECG performed during the study; however, a cardiologist considered these ECGs to be normal. No ECG results before enrollment were available. The patient had normal BP and HR at the beginning and throughout the trial. Before treatment, cholesterol level was 6.92 mmol/L (reference range = 3.88-6.83) and creatine phosphokinase was 249 U/L (reference range = 18-198). There was no history of smoking, substance abuse, angina, myocardial infarction, arrhythmia, hypercholesterolemia, or cardiovascular disease. The patient was not taking cardiovascular medications; however, it was not reported if the patient was taking other concomitant medications.
Treatment-Emergent Adverse Events
Approximately 80% of patients experienced ≥1 TEAE prerandomisation. Events reported with ≥5% incidence are shown in Table 2. Of the 2011 patients in the safety sample, 1614 reported a TEAE. Of these, 30.4%, 38.0%, and 11.8% reported a maximum severity of mild, moderate, and severe, respectively. One patient reported "more severe than baseline." Nausea, reported by 27.4% of patients, was the most frequent TEAE; only, 2.4% of all patients discontinued because of nausea (Figure 2). This equates to a discontinuation rate of 8.7% in patients experiencing nausea during atomoxetine treatment.
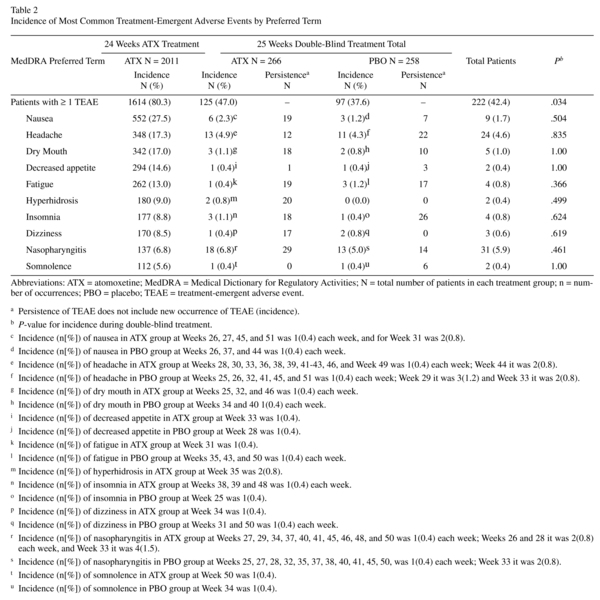
Postrandomisation, approximately 42.4% of patients experienced ≥1 TEAE(s), with the overall incidence of patients being significantly higher for atomoxetine than placebo (Table 2; P= .034); however, no significant differences were observed for any individual TEAE in any system organ class. Of the 524 randomized patients, 222 had a TEAE. Of these, 17.7%, 19.7%, and 4.4% reported a maximum severity of mild, moderate, and severe, respectively. Three patients reported "more severe than baseline." Post hoc summaries of the weekly incidence of the most common TEAEs showed few new occurrences of each respective TEAE for any given week. Persistence of TEAEs was numerically higher in atomoxetine vs. placebo for nausea, dry mouth, fatigue, hyperhidrosis, dizziness, and nasopharyngitis. It should be noted, however, for TEAEs such as fatigue, all 19 persistent occurrences took place in a single patient. Likewise, for dry mouth, all 17 persistent occurrences were for 3 patients.
Vital Signs and Body Weight
Prerandomisation, increases from baseline in SBP, DBP, and HR were significant, as were decreases from baseline for body mass index (BMI) and weight (Table 3; P<0.001 each). Frequencies of treatment-emergent abnormal high and low vital signs were as follows: SBP (1.3% and 0.7%), DBP (2.9% and 0.6%), and HR (0.5% and 0.2%). Only 1.7% and 5.2% of patients had abnormal weight gain or loss, respectively. The number (%) of patients with cardiac or weight-related TEAEs leading to discontinuation was small (tachycardia [0.4%], hypertension [0.3%], palpitations [0.1%], decreased appetite [0.1%], and BP increased [0.0%; 1 in 2011 patients]).
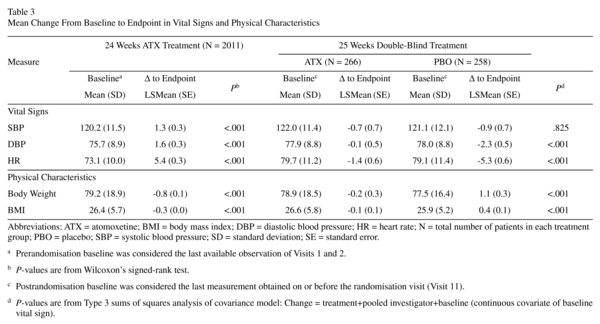
After discontinuation of atomoxetine (during the double-blind randomized withdrawal phase), patients on placebo showed a statistically significantly larger reduction in DBP and HR compared to patients who continued taking atomoxetine (Table 3; P<0.001 for each). The reductions in BMI and weight for atomoxetine were significantly different from the elevations observed for placebo (P<0.001 for each). Treatment-emergent abnormal low or high changes in SBP and DBP, HR, and weight were not significantly different between treatments. The number (%) of patients with cardiac or weight-related TEAEs leading to discontinuation was small for atomoxetine (hypertension [0.4%] myocardial infarction [0.4%]) and for placebo (myocardial infarction [0.4%] and weight gain [0.4%]).
Electrocardiograms
Prerandomisation changes in QTcB, QTcF, uncorrected QT, PR, QRS, and HR are shown in Table 4. No patient had a QTcF or QTcB >500 ms. Increases from baseline in QTcF and QTcB >30 ms were observed in 2.1% and 13.7% of patients, respectively. No patient showed an increase from baseline in QTcF and QTcB >60 ms. There was no shift to prolonged QTcB and QTcF (>500 ms) for patients with normal ECG parameters at baseline. For patients with borderline parameters at baseline, there was no shift to prolonged QTcB or QTcF (>500 ms).
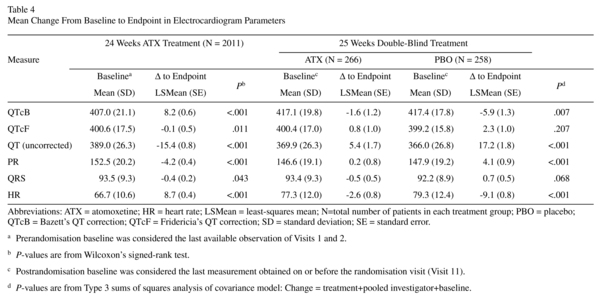
Postrandomisation changes in QTcB, QTcF, uncorrected QT, PR, QRS, and HR are shown in Table 4. No patient had a QTcF or QTcB >500 ms. There were no significant differences between atomoxetine and placebo in the frequencies of patients showing an increase from baseline in QTcF (1.4% vs. 2.6%) or QTcB (2.8% vs. 2.6%) >30 ms. No patients showed an increase from baseline in QTcF or QTcB >60 ms. There was no shift to prolonged QTcB or QTcF for patients with normal ECG parameters at baseline. For patients with borderline parameters at baseline, there was no shift to prolonged QTcB or QTcF (>500 ms).
Columbia Suicide-Severity Rating Scale and Suicide-Related Events
Prerandomisation, 56 of 2011 (2.8%) patients reported suicide-related events as solicited by the C-SSRS: 55 (2.7%) reported suicidal ideation and 2 (0.1%) reported suicidal behaviors, both of which were nonfatal suicide attempts. One single patient reported both suicidal ideation and behaviors. A subset (35 of 56) of patients reported a suicide-related event that was a new occurrence following enrollment or an event which worsened after enrollment. Only 10 (0.5%) patients, however, experienced an unsolicited TEAE of suicide ideation; 3 were SAEs, 2 of which led to study discontinuation. There were 135 (6.7%) patients who showed an improvement in suicidal ideation at endpoint relative to baseline.
The relative postrandomisation frequencies of suicide-related events, as solicited by the C-SSRS, were not significantly different between atomoxetine and placebo (6 of 266 [2.3%] vs. 3 of 258 [1.2%]; P= 0.504). Of the 6 atomoxetine-treated patients, 4 reported experiencing suicidal ideation, 1 reported experiencing suicidal behavior, and 1 reported experiencing both suicidal ideation and behavior. Of the 3 placebo-treated patients reporting suicide-related events, all 3 reported experiencing suicidal ideation and none reported experiencing suicidal behavior. Six atomoxetine-treated patients and 2 placebo-treated patients reported a suicide-related event that was a new occurrence following enrollment or an event which worsened after enrollment; however, there were no unsolicited TEAEs, SAEs, or discontinuations due to a suicide-related event. The relative frequencies of these new/worsened occurrences solicited via the C-SSRS were not significantly different between treatments.
Hamilton Anxiety Rating Scale-14 Items and Depression Rating Scale-17 Items
Prerandomisation changes from baseline in the HAMA and HAMD-17 total scores (-0.8 and -0.3) were statistically significant, but the differences were too small to be clinically relevant. Postrandomisation changes from baseline in the HAMA and HAMD-17 total scores were not significantly different between treatments.
Discussion
This study extends the findings of previous studies of atomoxetine in adults8,10,11,24,25 by providing evidence of the long-term safety of atomoxetine in the largest (to our knowledge) clinical trial of an ADHD medication (pediatric or adult) performed to date. The data demonstrate that atomoxetine, at the therapeutic dose studied, has an acceptable safety profile in a large international, multicenter sample of adult patients with ADHD. Adverse events reported at the highest rate occurred during the first 24 weeks of atomoxetine treatment (12-week open label and 12-week double-blind maintenance periods), when significant amelioration of ADHD symptoms was reported by Upadhyaya et al.26 to have occurred. These results were consistent with other short-term10,11,24,25 and long-term studies8 of atomoxetine in adults.
Nausea was the most frequent TEAE and the main reason for discontinuation prerandomisation; however, approximately 91.3% of patients who experienced nausea with atomoxetine remained in the study. This is likely the result of the typical characteristics of nausea; it occurred early in treatment, was usually mild or moderate, and tended to resolve rapidly in most patients. To that extent, all TEAEs appeared earlier in treatment, as shown by higher prerandomisation incidence rates relative to lower postrandomisation rates; the majority were mild or moderate in severity. A single death occurred during the clinical trial; this death was reported as suspected myocardial infarction by the principal investigator at the study site. The right-bundle branch block and baseline creatine phosphokinase and cholesterol values are not substantial risk factors for myocardial infarction in a 38-year-old male. Myocardial infarction would actually seem rather unlikely to be the cause of death and no conclusions can be drawn regarding possible causality.
Safety and tolerability are critical features in the profile of an ADHD medication. Many patients are required to take ADHD medication chronically. As a result, the nature of AEs often weighs heavily in patients risk-benefit assessment and may lead to noncompliance and/or discontinuation. In the current study, prerandomisation rates of discontinuation due to each respective AE were relatively low, and the impact of these TEAEs on initial patient compliance and overall tolerability was modest relative to other long-term studies of atomoxetine with similar dosing schedules8. Prerandomisation rates of discontinuation were somewhat higher, however, than those observed in short-term studies of atomoxetine, in which doses were adjusted based on tolerability10. During double-blind treatment, no single AE reported as reason for discontinuation was significantly different between treatments. The implication of these results is that education regarding the nonprogressive and transient nature of common TEAEs in clinical practice is important and may increase the number of patients that continue atomoxetine treatment.
Many ADHD medications have potential adverse cardiovascular effects including increases in BP and HR35. In the current study, increases in BP and HR observed after 24 weeks of atomoxetine treatment were consistent with a noradrenergically mediated increase in autonomic tone, but for most patients these increases were modest and asymptomatic. Indeed, less than 0.5% of patients discontinued atomoxetine due to treatment-emergent hypertension or tachycardia. These results are comparable with those in most short-term studies of atomoxetine in children18-20 and adults10,29,35,36. In patients receiving atomoxetine for an additional 25 weeks of double-blind treatment, BP and HR slightly declined relative to baseline at randomisation (Week 11). The declines in atomoxetine-treated patients, however, were significantly smaller than the declines observed in placebo-treated patients, whose BP and HR at endpoint returned to prerandomisation baseline values. The small percentages of treatment-emergent abnormally high or low BP and HR were not significantly different between the treatments. These results collectively extend the findings from previous studies, which have shown that increases in BP and HR usually occur within the first 2 months after atomoxetine treatment and are less likely to occur after 6 months of treatment10,18.
All ECG changes in the current study were expected based on HR changes and did not suggest an arrhythmogenic potential for atomoxetine. Mean change to endpoint for all ECG parameters was unremarkable including those for QT corrections. The observed categorical changes for QT corrections were also within acceptable limits. Taken together, these BP and ECG results did not identify any significant cardiovascular risks in this adult population after up to 1 year of treatment with atomoxetine beyond those already known; clinicians should consider the current prescribing information on cardiovascular risks for atomoxetine, and evaluate and monitor each individual patient accordingly37.
Atomoxetine has been associated with weight loss during treatment in children18-20 and adults8,29. In the current study, body weight decreased after 24 weeks of atomoxetine treatment, but appeared to plateau after an additional 25 weeks of treatment, despite significant differences at the 25-week time point between atomoxetine and placebo. The difference from placebo may have been magnified by a rebound in weight gain in those randomized to placebo; however, only 1 placebo-treated patient discontinued during double-blind treatment due to weight gain. These results are comparable with results of a long-term study of atomoxetine in adults8 and are consistent with findings in children38.
Patients with ADHD are at greater risk for suicidal behavior than the general public39. Whether this increased risk is due to the impulsivity inherent in ADHD, other comorbid psychiatric illnesses, or to other factors that have yet to be identified is unknown. No patient in atomoxetine ADHD trials has committed suicide, and high rates of suicide ideation in both children and adults are uncommon. However, small, but significantly higher rates of suicidal ideation have been reported in a pediatric ADHD population treated with atomoxetine vs. placebo40. In the current study, CSSR-S results from double-blind treatment did not suggest a link between atomoxetine and suicidality; there were no significant differences between treatments, and no randomized patient discontinued due to a suicide-related event. Furthermore, there were no significant differences between treatments in the HAMD and HAMA, supporting the hypothesis that atomoxetine did not negatively impact depression or anxiety, respectively.
Several factors limit the interpretation of the data presented here. No comparisons with other drugs were included. The ability to generalize these results to typical outpatients is somewhat limited, as patients in this study had relatively fewer comorbid medical conditions and concomitant medications than their general outpatient counterparts. Furthermore, the low rate of AEs and discontinuations during double-blind treatment may be artifacts of the fact that all patients in the double-blind treatment phase of this study had been treated with atomoxetine for 24 weeks before randomisation. Indeed, these data only describe a subpopulation that responded to atomoxetine and were compliant with the lengthy study protocol schedule.
This study demonstrated that atomoxetine exhibited an acceptable safety profile in adults with ADHD during 24 weeks of treatment and during an additional 25 weeks of double-blind treatment in the largest clinical trial of ADHD in adults to date. No clinically meaningful rebound effects related to safety were seen after abrupt discontinuation of atomoxetine, suggesting that patients do not need to be tapered when discontinuing treatment. Patients and their physicians will need to balance the value of an individual's treatment against the risk and management of potential side effects. Individuals that achieve an acceptable level of improvement in ADHD symptoms and atomoxetine tolerability are those who should consider continued medical treatment.
References
1. Biederman J, Spencer T. Attention-deficit/hyperactivity disorder (ADHD) as a noradrenergic disorder. Biol Psychiatry 1999; 46(9): 1234-1242. [ Links ]
2. Faraone SV, Biederman J. Neurobiology of attention-deficit hyperactivity disorder. Biol Psychiatry 1998; 44(10): 951-958. [ Links ]
3. Zametkin AJ, Liotta W. The neurobiology of attention-deficit/hyperactivity disorder. J Clin Psychiatry 1998; 59(Suppl 7): 17-23. [ Links ]
4. Barkley RA, Fischer M, Smallish L, Fletcher K. The persistence of attention deficit/hyperactivity disorder into young adulthood as a function of reporting source and definition of disorder. J Abnorm Psychol 2002; 111(2): 279-289. [ Links ]
5. Mannuzza S, Klein RG, Moulton JL 3rd. Persistence of attention-deficit/hyperactivity disorder into adulthood: what have we learned from the prospective follow-up studies? J Atten Disord 2003; 7(2): 93-100. [ Links ]
6. Adler L, Cohen J. Diagnosis and evaluation of adults with attention-deficit/hyperactivity disorder. Psychiatr Clin North Am 2004; 27(2): 187-201. [ Links ]
7. Kooij SJ, Bejerot S, Blackwell A, Caci H, Casas-Brugué M, Carpentier PJ, et al. European consensus statement on diagnosis and treatment of adult ADHD: the European network adult ADHD. BMC Psychiatry 2010; 10:67. [ Links ]
8. Adler LA, Spencer T, Brown TE, Holdnack J, Saylor K, Schuh K, et al. Once-daily atomoxetine for adult attention-deficit/hyperactivity disorder: a 6-month, double-blind trial. J Clin Psychopharmacol 2009; 29(1): 44-50. [ Links ]
9. Biederman J, Mick E, Surman C, Doyle R, Hammerness P, Kotarski M, et al. A randomized, 3-phase, 34-week, double-blind, long-term efficacy study of osmotic-release oral system methylphenidate in adults with attention-deficit/ hyperactivity disorder. J Clin Psychopharmacol 2010; 30(5): 549-553. [ Links ]
10. Michelson D, Adler L, Spencer T, Reimherr FW, West SA, Allen AJ, et al. Atomoxetine in adults with ADHD: two randomized, placebo-controlled studies. Biol Psychiatry 2003; 53(2): 112-120. [ Links ]
11. Spencer T, Biederman J, Wilens T, Prince J, Hatch M, Jones J, et al. Effectiveness and tolerability of tomoxetine in adults with attention deficit hyperactivity disorder. Am J Psychiatry 1998; 155(5): 693-695. [ Links ]
12. Spencer T, Biederman J, Wilens T, Doyle R, Surman C, Prince J, et al. A large, double-blind, randomized clinical trial of methylphenidate in the treatment of adults with attention deficit/hyperactivity disorder. Biol Psychiatry 2005; 57(5): 456-463. [ Links ]
13. Bouffard R, Hechtman L, Minde K, Iaboni-Kassab F. The efficacy of 2 different dosages of methylphenidate in treating adults with attention-deficit hyperactivity disorder. Can J Psychiatry 2003; 48(8): 546-554. [ Links ]
14. Kooij JJ, Burger H, Boonstra AM, Van der Linden PD, Kalma LE, Buitelaar JK. Efficacy and safety of methylphe-nidate in 45 adults with attentiondeficit/hyperactivity disorder. A randomized placebo-controlled double-blind cross-over trial. Psychol Med 2004; 34(6): 973-982. [ Links ]
15. Medori R, Ramos-Quiroga JA, Casas M, Kooij JJ, Niemelä A, Trott GE, et al. A randomized, placebo-controlled trial of three fixed dosages of prolonged-release OROS methylphenidate in adults with attention-deficit/hyperactivity disorder. Biol Psychiatry 2008; 63(10): 981-989. [ Links ]
16. Himpel S, Banaschewski T, Heise CA, Rothenberger A. The safety of non-stimulant agents for the treatment of attention-deficit hyperactivity disorder. Expert Opin Drug Saf 2005; 4(2): 311-321. [ Links ]
17. Vaughan B, Fegert J, Kratochvil CJ. Update on atomoxetine in the treatment of attention-deficit/hyperactivity disorder. Expert Opin Pharmacother 2009; 10(4): 669-676. [ Links ]
18. Kelsey DK, Sumner CR, Casat CD, Coury DL, Quintana H, Saylor KE, et al. Once-daily atomoxetine treatment for children with attention-deficit/hyperactivity disorder, including an assessment of evening and morning behavior: a double-blind, placebo-controlled trial. Pediatrics 2004; 114(1): e1-e8. [ Links ]
19. Michelson D, Faries D, Wernicke J, Kelsey D, Kendrick K, Sallee FR, et al. Atomoxetine ADHD Study Group. Atomoxetine in the treatment of children and adolescents with attention-deficit/hyperactivity disorder: a randomized, placebo-controlled, dose-response study. Pediatrics 2001; 108(5): E83. [ Links ]
20. Michelson D, Allen AJ, Busner J, Casat C, Dunn D, Kratochvil C, et al. Once-daily atomoxetine treatment for children and adolescents with attention deficit hyperactivity disorder: a randomized, placebo-controlled study. Am J Psychiatry 2002; 159(11): 1896-901. [ Links ]
21. Michelson D, Buitelaar JK, Danckaerts M, Gillberg C, Spencer TJ, Zuddas A, et al. Relapse prevention in pediatric patients with ADHD treated with atomoxetine: a randomized, double-blind, placebo-controlled study. J Am Acad Child Adolesc Psychiatry 2004; 43(7): 896-904. [ Links ]
22. Newcorn JH, Michelson D, Kratochvil CJ, Allen AJ, Ruff DD, Moore RJ. Atomoxetine Low-dose Study Group. Low-dose atomoxetine for maintenance treatment of attention-deficit/hyperactivity disorder. Pediatrics 2006; 118(6): e1701-e1706. [ Links ]
23. Spencer T, Heiligenstein JH, Biederman J, Faries DE, Kratochvil CJ, Conners CK, et al. Results from 2 proof-of-concept, placebo-controlled studies of atomoxetine in children with attention-deficit/hyperactivity disorder. J Clin Psychiatry 2002; 63(12): 1140-1147. [ Links ]
24. Wilens TE, Adler LA, Weiss MD, Michelson D, Ramsey JL, Moore RJ, et al. Atomoxetine treatment of adults with ADHD and comorbid alcohol use disorders. Drug Alcohol Depend 2008; 96(1-2): 145-154. [ Links ]
25. Adler LA, Liebowitz M, Kronenberger W, Qiao M, Rubin R, Hollandbeck M, et al. Atomoxetine treatment in adults with attention-deficit/hyperactivity disorder and comorbid social anxiety disorder. Depress Anxiety 2009; 26(3): 212-221. [ Links ]
26. Upadhyaya H, Ramos-Quiroga JA, Adler JA, Wi-lliams D, Tanaka Y, Lane JR, et al. Maintenance of Response after Open-Label Treatment with Atomoxetine Hydrochloride in International European and Non-European Adult Outpatients with Attention-Deficit/Hyperactivity Disorder: A Placebo-Controlled, Randomised Withdrawal Study. Eur. J Psychiat. Submitted 2012. [ Links ]
27. Conners CK, Erhardt D, Sparrow E. Conners' Adult ADHD Rating Scales (CAARS). North Tonawanda: Multi-Health Systems, Inc.; 1999. [ Links ]
28. Guy W. ECDEU Assessment Manual for Psychopharmacology, Revised. Bethesda, MD: US Department of Health, Education, and Welfare; 1976. [ Links ]
29. Adler LA, Wilens T, Zhang S, Dittmann RW, D'Souza DN, Schuh L, et al. Atomoxetine treatment outcomes in adolescents and young adults with attention-deficit/hyperactivity disorder: results from a post hoc, pooled analysis. Clin Ther 2012; 34(2): 363-373. [ Links ]
30. Posner K, Oquendo MA, Gould M, Stanley B, Davies M. Columbia Classification Algorithm of Suicide Assessment (C-CASA): classification of suicidal events in the FDA's pediatric suicidal risk analysis of antidepressants. Am J Psychiatry 2007; 164(7): 1035-1043. [ Links ]
31. Hamilton M. The assessment of anxiety states by rating. Br J Med Psychol 1959; 32(1): 50-55. [ Links ]
32. Riskind JH, Beck AT, Brown G, Steer RA. Taking the measure of anxiety and depression. Validity of the reconstructed Hamilton scales. J Nerv Ment Dis 1987; 175(8): 474-479. [ Links ]
33. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry 1960; 23: 56-62. [ Links ]
34. Hamilton M. Development of a rating scale for primary depressive illness. Br J Soc Clin Psychol 1967; 6(4): 278-296. [ Links ]
35. Stiefel G, Besag FM. Cardiovascular effects of methylphenidate, amphetamines and atomoxetine in the treatment of attention-deficit hyperactivity disorder. Drug Saf 2010; 33(10): 821-842. [ Links ]
36. Simpson D, Plosker GL. Atomoxetine: a review of its use in adults with attention deficit hyperactivity disorder. Drugs 2004; 64(2):205-222. [ Links ]
37. Strattera (atomoxetine hydrochloride) (package insert). Indianapolis: Eli Lilly and Company; 2011. [ Links ]
38. Spencer TJ, Kratochvil CJ, Sangal RB, Saylor KE, Bailey CE, Dunn DW, et al. Effects of atomoxetine on growth in children with attention-deficit/hyperactivity disorder following up to five years of treatment. J Child Adolesc Psychopharmacol 2007; 17(5): 689-700. [ Links ]
39. Impey M, Heun R. Completed suicide, ideation and attempt in attention deficit hyperactivity disorder. Acta Psychiatr Scand 2012; 125(2): 93-102. [ Links ]
40. Bangs ME, Tauscher-Wisniewski S, Polzer J, Zhang S, Acharya N, Desaiah D, et al. Meta-analysis of suicide-related behavior events in patients treated with atomoxetine. J Am Acad Child Adolesc Psychiatry 2008; 47(2): 209-218. [ Links ]
![]() Correspondence:
Correspondence:
Himanshu Upadhyaya, MBBS, MS
Senior Medical Advisor
ADAP Team, Neuroscience Platform
Eli Lilly & Co
Lilly Corp. Center, D/C 6112
Indianapolis, IN 46285
Phone: 317-655-3516
Fax: 713-592-0916
Received: 14 August 2012
Revised: 28 March 2013
Accepted: 17 April 2013

