










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versión impresa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.81 no.5 may. 2006
COMUNICACIÓN CORTA
Asociación de albinismo oculocutáneo y distrofia granular en una familia
Familiar case of granular dystrophy and oculocutaneous albinism
Gómez-Valcárcel M.1, Chin-Wong J.L.1, Álvarez-Verduzco O.1, Niño-Pecina A.2, Villanueva-Mendoza C.1
Departamento de Genética. Hospital Dr. Luis Sánchez Bulnes. Asociación para Evitar la Ceguera en México, IAP. México.
1 Doctor en Medicina. Asociación para Evitar la Ceguera en México (APEC).
2 Doctor en Medicina. Centro Médico de Toluca. México.
Dirección para correspondencia
RESUMEN
Caso clínico: Paciente femenino de 35 años de edad con mala agudeza visual desde la infancia. A la exploración se encontró baja agudeza visual, nistagmo, hipopigmentación de piel y cabello amarillento, córnea con depósitos blanquecinos en estroma central y anterior, transiluminación de iris e hipoplasia foveal. Se diagnosticaron albinismo oculocutáneo y distrofia corneal granular. Se encontró albinismo oculocutáneo en dos hermanos y distrofia granular en tres hermanos, la madre y el hijo.
Discusión: La distrofia corneal granular se transmite genéticamente siguiendo un patrón autosómico dominante e independiente del albinismo oculocutáneo. Este es el primer caso publicado de presentación concomitante de ambas entidades.
Palabras clave: Albinismo oculocutáneo, distrofia granular.
ABSTRACT
Clinical case: A 35-year-old female patient with blurred vision since childhood, for which no treatment had been given, presented with poor visual acuity. She had white skin and fair yellow hair. There were several well circumscribed deposits in the central and anterior corneal stroma, and iris transillumination and foveal hypoplasia were evident. The clinical diagnosis was oculo-cutaneous albinism and granular corneal dystrophy. We found oculo-cutaneous albinism in two brothers and granular dystrophy in three brothers, the mother and a son.
Discussion: Corneal dystrophy is an autosomal dominant disorder inherited independently of oculocutaneous albinism, which is inherited as an autosomal recessive condition. This is the first case report of granular dystrophy concurrent with oculocutaneous albinism (Arch Soc Esp Oftalmol 2006; 81: 289-292).
Key words: Oculocutaneous albinism, granular dystrophy.
Introducción
La distrofia granular (DG) o Groenouw tipo I, es una patología corneal bilateral que se caracteriza por la presencia de opacidades blanquecinas, discretas, de forma variable, en el estroma anterior y central. Se inicia en la primera o segunda décadas de la vida. Con la evolución de la enfermedad, las lesiones crecen en número y tamaño y empiezan a coalescer; el estroma entre opacidades permanece transparente pero posteriormente toma aspecto de vidrio esmerilado. Las alteraciones visuales son raras antes de la quinta década de la vida; sin embargo, en algunos pacientes pueden producirse erosiones epiteliales e incremento en la opacidad corneal, con deterioro importante en la agudeza visual.
El albinismo es un grupo heterogéneo de patologías hereditarias caracterizadas por anormalidades en la síntesis de melanina asociada a cambios en el desarrollo del sistema visual. El albinismo oculocutáneo (AOC) involucra ojos, piel y cabello. Los signos oftalmológicos son variables, e incluyen nistagmo, hipopigmentación del tracto uveal y del epitelio pigmentado de la retina, transiluminación del iris, hipoplasia foveal y decusación anormal de las fibras del nervio óptico a nivel del quiasma (1).
Se presenta un caso con manifestación simultánea de DG y AOC.
Caso Clínico
Mujer de 35 años acude a consulta por mala agudeza visual (AV) desde la infancia, sin tratamiento. A la exploración se encontró cabello amarillento, al igual que cejas y pestañas; piel rojiza, sin nevos (fig. 1); agudeza visual con corrección de 20/400 (–9,50 –5,50 x 135º) en ojo derecho (OD) y 20/200 (–6,50 –5,50 x 20º) en ojo izquierdo (OI); exotropía de 50 dioptrías, nistagmo horizontal; ambos ojos (AO) con presión intraocular de 12 mmHg, conjuntiva normal, córnea con algunos depósitos estromales blanquecinos centrales y anteriores bien circunscritos (fig. 2), cámara anterior sin células ni flare, iris claro con transiluminación, cristalino transparente e hipoplasia foveal. Con los datos clínicos oculares y sistémicos se establecieron los diagnósticos de AOC y DG y se citó a la familia para valoración clínica.

Fig. 1. Fotografía clínica de la paciente demostrando las
características de hipopigmentación de cabello y piel.
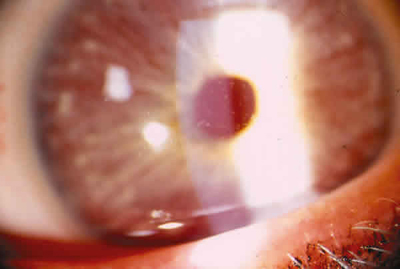

Fig. 2. Fotografía clínica que muestra depósitos estromales
en "miga de pan". a) Ojo derecho. b) Ojo izquierdo.
Se trata de una familia de nueve miembros, con padres de 74 y 64 años. No hay consanguinidad y proceden de localidades diferentes. La DG está presente en la madre, tres hermanos y su hijo, mientras que el AOC aparece en dos de los hermanos pero sin datos de DG (fig. 3).

Fig. 3. Árbol genealógico.
Discusión
La distrofia granular es la distrofia estromal más común y se transmite con herencia autosómica dominante. Se produce por mutación en el gen big-h3 del cromosoma 5q31 que codifica la molécula de queratoepitelina.
En la mayoría de los casos, el tratamiento se limita a corrección del defecto refractivo. En algunos pacientes puede haber erosiones epiteliales recurrentes que requieren empleo de lentes de contacto terapéuticos y lágrimas artificiales. Cuando la agudeza visual está fuertemente afectada, puede considerarse el tratamiento quirúrgico, de acuerdo a la profundidad y extensión de las lesiones.
El tratamiento con queratoplastía penetrante se realiza en 4.1 a 37,7% de los casos, con una tasa de recurrencia hasta del 87% (2), que requiere una nueva intervención (3).
Se ha empleado también la queratotomía fototerapéutica para el tratamiento de esta distrofia (4).
El AOC es un grupo de enfermedades caracterizado por reducción en la síntesis de melanina. Si la síntesis de melanina es deficiente en la piel, cabello y ojos se usa el término de AOC, si sólo ocurre a nivel ocular se considera como albinismo ocular. Existen al menos diez variantes de AOC que han sido subclasificadas en dos categorías de acuerdo a la presencia o ausencia de actividad de tirosinasa en los melanocitos: tirosinasa-negativo (AOC 1) y tirosinasa-positivo (AOC 2). El patrón de herencia es autosómico recesivo. El AOC 1 es la forma más severa de albinismo y se produce por alteraciones en el gen de la tirosinasa, que se encuentra en el cromosoma 11q14-21. En el AOC 2 hay producción de feomelanina y la mutación se localiza en el cromosoma 15q11.2-q12 (5).
Los datos que se observan en la paciente como mala agudeza visual, nistagmo, ausencia de nevos, hipopigmentación de piel, cabello y ojos indican que se trata de AOC 1.
Las características clínicas de la paciente sugieren que la mala agudeza visual se debe en mayor medida al albinismo oculocutáneo, por lo que el tratamiento se limita en este momento a corrección del defecto refractivo. Para la distrofia corneal los datos familiares indican un patrón autosómico dominante, de manera independiente a la patología de albinismo oculocutáneo. Es interesante la asociación de las dos patologías en una familia considerándose como eventos independientes, ya que hasta el momento no se ha encontrado una relación de los genes involucrados y los loci conocidos para estas patologías se ubican en diferentes cromosomas. Hasta nuestro conocimiento, ésta es la primera publicación de presentación concomitante de ambas entidades.
Bibliografía
1. Young TL. Ophthalmic genetics/inherited eye disease. Curr Opin Ophthalmol 2003; 14: 296-303. [ Links ]
2. Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea 2003; 22: 19-21. [ Links ]
3. Lyons CJ, McCartney AC, Kirkness CM, Ficker LA, Steele AD, Rice NS. Granular corneal dystrophy. Visual results and pattern of recurrence after lamellar or penetrating keratoplasty. Ophthalmology 1994; 101: 1812-1817. [ Links ]
4. Seitz B, Behrens A, Fischer M, Langenbucher A, Naumann GO. Morphometric analysis of deposits in granular and lattice corneal dystrophy. Histopathologic implications for phototherapeutic keratectomy. Cornea 2004; 23: 380-385. [ Links ]
5. MacDonald IM, Tran M, Musarella MA. Ocular Genetics: Current Understanding. Surv Ophthalmol 2004; 49: 159-196. [ Links ]
![]() Dirección para correspondencia
Dirección para correspondencia
María Gómez Valcárcel
Hospital Dr. Luis SánchezBulnes
Asociación para Evitar la Ceguera en México
C/ Vicente García Torres Nº 46
Col. San Lucas Coyoacán
04030 México D.F.
México
E-mail: maria_01_05@hotmail.com
Recibido: 5.5.05
Aceptado:18.5.06

