










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.96 no.4 Madrid abr. 2004
|
PUNTO DE VISTA |
Carcinogénesis gástrica
S. de la Riva, M. Muñoz-Navas y J. J. Sola1
Servicios de Aparato Digestivo y 1Anatomía Patológica. Clínica Universitaria de Navarra. Pamplona
1. INTRODUCCIÓN
El adenocarcinoma gástrico continúa siendo en la actualidad un importante problema sanitario a nivel mundial, ocupando el segundo lugar en frecuencia de los tumores malignos (1). Aunque de forma global su incidencia parece haber disminuido, tanto su frecuencia como su tasa de mortalidad muestran una importante variación geográfica. En los países orientales, este de Europa y sur de América la incidencia de esta enfermedad alcanza rangos epidémicos y constituye la primera causa de muerte por tumores malignos (2). Por el contrario en otras regiones geográficas la incidencia de esta enfermedad es baja (América del Norte, Oeste de Europa, Australia, Nueva Zelanda e Israel). Estas diferencias han sido en parte atribuidas a factores ambientales como alimenticios, referidos a la conservación y preparación de los alimentos, e infecciosos, en relación con la incidencia de infección por H. pylori en la población. Tal es la posible influencia de estos factores, que la progresiva disminución en la tasa de incidencia y de mortalidad de esta enfermedad observada desde la década de los 30 hasta los últimos 10 años, se ha atribuido a los cambios dietéticos y de conservación de los alimentos (3-5).
No obstante este cambio en los factores ambientales no explica por completo las variaciónes geográficas conocidas ni la diferente evolución y pronóstico de la enfermedad.
Actualmente, se acepta que la carcinogénesis gástrica es un proceso progresivo en el que intervienen múltiples factores tanto ambientales y epidemiológicos como genéticos, y cuya interacción parece influir no sólo en el desarrollo sino también en la progresión de la enfermedad (6).
2. FACTORES CARCINOGÉNICOS
2.1. Factores ambientales
En los últimos años y puesto que el cáncer gástrico continúa siendo uno de los tumores malignos más frecuentes en la población mundial muchos estudios han centrado su atención en identificar factores de riesgo ambientales que justifiquen su amplia variación geográfica.
Factores alimenticios
Las poblaciones con un alto consumo de sal, alimentos ahumados, picantes o grasas fritas presentan una elevada tasa de incidencia de adenocarcinoma gástrico indicando su posible papel en la carcinogénesis (5,7-10).
Alimentos o agua rica en nitritos, la alta ingesta de carbohidratos y el poco acceso y consumo de fruta, verdura fresca, leche, vitaminas A, C y E y selenio parecen incrementar el riesgo de desarrollar cáncer gástrico (3,5).
Los factores clásicamente considerados carcinogénicos como el tabaco, el alcohol o la toma de te verde, presentan todavía en la literatura resultados poco consistentes (6,11).
Cabe destacar en la actualidad, el incremento progresivo en los países industrializados del adenocarcinoma gástrico de localización cardial frente al predominio de los distales y prácticamente la ausencia de los proximales en los países en vías de desarrollo (9,10). Estas diferencias se atribuyen a una mayor incidencia de la enfermedad por reflujo gastro-esofágico en los países desarrollados en relación con la dieta y el aumento de la tasa de obesidad. También el consumo de forma crónica de antisecretores tipo inhibidores de la bomba de protones favorece la transformación de los nitratos de la saliva a nitritos, agentes que favorecen la enfermedad (12).
Infección por H. pylori
A comienzos de los años 80 se descubrió en las biopsias de la mucosa gástrica de pacientes con gastritis y ulcus péptico la presencia de una bacteria que inicialmente fue denominada Campylobacter piloridis (13) y posteriormente C. pylori. En la actualidad se denomina Helicobacter pylori. Se trata de una bacteria con forma de espiral que pertenece al grupo de los gérmenes gram-negativos y que utiliza una gran variedad de estrategias para lograr sobrevivir en el medio ácido.
La infección crónica por H. pylori conduce al desarrollo de una gastritis crónica, mediada por la activación de una red compleja de mediadores inflamatorios incluidos IL-8, citoquinas proinflamatorias (IL-1, IL-6, FNTα) y péptidos inmunosupresores (IL-10) (14).
A su vez esta inflamación crónica conduce a alteraciones en el ciclo celular favoreciendo la replicación de las células epiteliales, incrementando la tasa de apoptosis y aumentando la liberación de sustancias oxidantes. Todo esto en combinación con la depleción de las defensas antioxidantes predispone a la carcinogénesis gástrica por aumentar la probabilidad de mutaciones de ADN (14).
La acumulación de estas mutaciones puede conducir al desarrollo de lesiones premalignas e iniciar el proceso de metaplasia, displasia y adenocarcinoma gástrico (4, 15).
Actualmente la infección crónica por la bacteria H. pylori es considerada como carcinógeno del grupo I por la Organización Mundial de la Salud (OMS) (16) y parece jugar un importante papel en el desarrollo del adenocarcinoma gástrico distal.
Varios estudios clínicos y epidemiológicos han observado esta asociación estimándose que el menos el 1% de las infecciones por H. pylori pueden conducir al desarrollo del cáncer gástrico según la secuencia anteriormente expuesta (17), aumentando el riesgo de 2,7 a 12 veces el de la población general (18). De forma global el 8% de los tumores gástricos están relacionados causalmente por la infección por H. pylori (19).
Al igual que la incidencia de la enfermedad, la tasa poblacional de infección por H. pylori presenta una gran variación geográfica. En general áreas con alto riesgo de adenocarcinoma gástrico muestran altas tasas de infección por H. pylori (20,21).
Del mismo modo se ha recogido una incidencia de cáncer gástrico mayor en poblaciones con nivel socioeconómico bajo. Esta observación parece estar relacionada en parte con factores alimenticios como menor acceso y consumo de frutas y verduras frescas y/o una mayor tasa de infección por H. pylori adquirida a edades más tempranas (22).
En un intento de explicar la mayor asociación de la infección concomitante por H. pylori con el adenocarcinoma de tipo intestinal frente al tipo difuso, se ha propuesto la existencia de dos vías de actuación diferentes según el tipo histológico de cáncer gástrico, con progresivos cambios histológicos en el adenocarcinoma de tipo intestinal que no tienen lugar en el de tipo difuso (23) (Fig. 1).
Infección por virus del Epstein Barr (EBV)
Descubierto por Epstein en 1964, es un herpes-virus icosaédrico que contiene una doble cadena lineal de ADN. Fue relacionado por primera vez con el cáncer gástrico en 1990 tras observar mediante PCR e hibridación in situ la presencia de su material genético en pacientes con cáncer gástrico (24). Su asociación ha sido encontrada en numerosos estudios de diferentes regiones geográficas (25,26).
A diferencia del H. pylori, su infección ha sido observada por igual en el cáncer gástrico de tipo difuso que en el intestinal (27).
Todavía se desconoce cuál es su mecanismo carcinogénico. Incluso se ha propuesto la existencia de dos vías de carcinogénesis distintas en función de la presencia o ausencia de la infección concomitante por este virus (28).
2.2. Factores genéticos
En relación con la influencia de los factores genéticos en el desarrollo del adenocarcinoma gástrico, se ha observado que el cáncer gástrico puede estar presente en familias durante dos o tres generaciones y que además los miembros de una familia con antecedentes de adenocarcinoma gástrico tienen un riesgo incrementado en dos o incluso tres veces el de la población general, implicando una posible agregación familiar (29).
Sin embargo los estudios que incluyen a miembros de una misma familia son limitados y sus resultados se ven influidos por los factores ambientales, puesto que generalmente los miembros de una misma familia están bajo las mismas condiciones dietéticas y ambientales (29).
Una observación que implica de manera indirecta el posible papel de factores genéticos en la carcinogénesis gástrica, es la mayor asociación de esta enfermedad con el grupo sanguíneo A en comparación con los otros grupos sanguíneos. Esta asociación es más marcada en varones y en el tipo histológico difuso frente al intestinal (5, 9,29).
2.3. Condiciones precursoras
Determinados cambios histológicos de la mucosa gástrica sana aumentan significativamente el riesgo de desarrollar un adenocarcinoma gástrico.
Entre ellos cabe destacar:
Gastritis crónica atrófica
Lesión precancerosa que se encuentra presente en el 90% de los adenocarcinomas gástricos. En general requiere un largo periodo de evolución hasta el desarrollo del cáncer gástrico. En la mayoría de los estudios en los que el seguimiento de los pacientes fue superior a los 10 años, el riesgo de desarrollar cáncer gástrico fue de 1 por cada 150 pacientes por año, incrementándose este riesgo al 10% después de los 15 años de seguimiento (30,31).
Su mecanismo carcinogénico parece partir de la disminución de la secreción de ácido clorhídrico y pepsina, aumentando el pH gástrico, lo que favorece la proliferación de gérmenes reductores de los nitratos de la dieta. La formación de nitrosamidas y nitrosaminas junto con algunos factores dietéticos como son la ingesta excesiva de sal o la ingesta inadecuada de vegetales y fruta fresca pueden inducir mutaciones del ADN en las células epiteliales favoreciendo la aparición y progresión de cambios tisulares como metaplasia intestinal y displasia, considerados lesiones premalignas (4,32).
Anemia perniciosa
Condición que cursa con atrofia gástrica y que aumenta el riesgo de desarrollar cáncer gástrico (33) aunque sólo el 5-10% de estos pacientes lo desarrollan (31).
Gastrectomía parcial
Los pacientes con patología benigna sometidos a esta cirugía tienen un riesgo incrementado de desarrollar adenocarcinoma gástrico a partir de los 10 años de la intervención (34). Entre los 15 y 25 años tras el procedimiento el riesgo se incrementa un 50 y 70% respectivamente (31).
Enfermedad de Ménétrier (gastropatía hipertrófica)
El riesgo de desarrollar cáncer gástrico a partir de este cambio tisular es alto, situándose entre el 10-15% en algunas series, pero puesto que ya de por sí se trata de una condición extremadamente rara, este porcentaje de transformación resulta insignificante (35).
Pólipos adenomatosos
Los pólipos se encuentran con relativa frecuencia en la mucosa gástrica. Se clasifican en dos tipos:
-No neoplásicos: no presentan capacidad degenerativa (hiperplásicos, hamartomatosos, inflamatorios o heterotópicos).
-Neoplásicos: adenomas. Constituyen el 15-20% de los pólipos encontrados en la mucosa gástrica. Poseen potencial neoplásico con una incidencia de malignización que oscila entre el 5-15% de los adenoma tubulares y el 15-75% de los adenomas vellosos. La tendencia a la malignización está directamente relacionada con el tamaño del pólipo y con la presencia o grado de displasia (36).
Úlcera péptica
La posibilidad de transformación de una úlcera péptica benigna en maligna está todavía en discusión con opiniones discordantes al respecto. Aunque la mayoría de los autores niegan esta posibilidad, se debe tener en cuenta el papel que parece desarrollar la infección por H. pylori en el proceso de la carcinogénesis gástrica (37).
Esófago de Barrett
El aumento de la incidencia del adenocarcinoma gástrico cardial en los países indutrializados parece estrechamente relacionado con el aumento de la incidencia de la enfermedad por reflujo gastro-esofágico y del esófago de Barrett (38,39). Todavía son necesarios estudios más amplios para determinar otros factores que intervengan en su desarrollo y de esta manera poder establecer si realmente el cáncer gástrico proximal es una entidad con diferente etiopatogenia y evolución que el distal.
2.4. Factores moleculares
Aunque hay suficiente evidencia de que las alteraciones genéticas juegan un papel importante en el proceso multipaso de la carcinogénesis gástrica y en su progresión, el amplio número de factores analizados y los diferentes resultados obtenidos no permiten por el momento establecer conclusiones definitivas (9,40-42). Lo que sí parece claro es que la conversión de células normales gástricas en tumorales es un proceso lento y progresivo con acumulación de múltiples alteraciones moleculares, que se ha querido equiparar al proceso de carcinogénesis del cáncer colorrectal. Esta acumulación de cambios genéticos incluye mutaciones y/o amplificación-sobre-expresión de oncogenes (c-Ki-ras, c-erb-B2, K-sam, hst/int-2, c-met y c-myc), inactivación de genes supresores de tumor (p53, APC, DCC y RB1), y alteración de microsatélites (pérdida de heterozigosidad o inestabilidad de microsatélites) en una o más regiones cromosómicas como 1p, 5q, 7q, 12q, 13q, 17p, 18q e incluso en el cromosoma Y (40,43,44) (Tabla I).
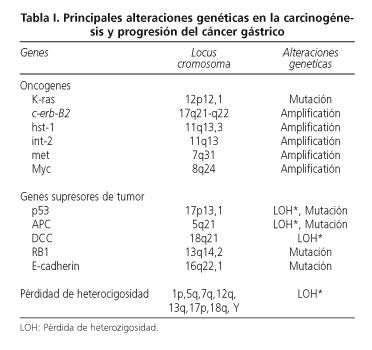
Los resultados de los diferentes estudios de análisis molecular en el cáncer gástrico han puesto de manifiesto la posible existencia de dos vías de carcinogénesis distintas en función del tipo histológico de adenocarcinoma difuso o intestinal (45-49). El cáncer gástrico de tipo intestinal parece tener relación con la existencia de cambios tisulares en la mucosa gástrica tipo metaplasia intestinal y presenta semejanzas con la evolución molecular del cancer colorrectal (47). Se desarrolla a través de un proceso que se inicia con la gastritis crónica y se continúa con la gastritis atrófica, la metaplasia intestinal y la displasia (4). Por otro lado parece que la historia natural del cáncer gástrico de tipo difuso prescinde de esta evolución multipaso (46,50-52).
3. MODELO ACTUAL DE CARCINOGÉNESIS
La clasificación histológica de Lauren (1965) del cáncer gástrico en difuso e intestinal es la más frecuentemente utilizada por la mayoría de los autores (53).
Estos dos tipos histológicos de cáncer gástrico probablemente reflejan no sólo las diferencias morfológicas que permiten su clasificación, sino también distintas características clínicas, epidemiológicas y patogénicas.
Por todo ello se ha propuesto la existencia de dos vías de carcinogénesis diferentes según el tipo histológico de cáncer gástrico con distinta influencia de los factores ambientales y variación en la presencia y predominio de las alteraciones moleculares.
El tipo intestinal, como ya se ha comentado anteriormente, parece desarrollarse a partir de sucesivos cambios tisulares de forma similar a la vía de carcinogénesis del cáncer colorrectal. O’Connor y cols. (54), Watanabe y cols. (24) y Correa (4) propusieron, en relación con los factores ambientales, un modelo de carcinogénesis gástrica en espiral, de manera que un acúmulo del número de los factores de riesgo ambientales suponía un tiempo de desarrollo más corto, y su disminución un alargamiento del tiempo de evolución (Fig. 2).
Además y del mismo modo que los autores anteriomente citados, Yasui y cols. (42) recogieron de la literatura todos los factores moleculares analizados e implicados en la carcinogénesis gástrica, en un intento de equiparar la carcinogénesis gástrica al proceso carcinogénico colorrectal. Debido a las diferencias observadas entre los dos tipos histológicos de adenocarcinoma gástrico, plantearon la posibilidad de la existencia de dos vías de desarrollo distintas con la presencia y predominio de diferentes alteraciones moleculares (Fig. 3).
Lo que sí parece claro es que en la carcinogénesis del adenocarcinoma gástrico de tipo intestinal acontecen una serie de cambios tisulares que pueden progresar hacia la malignidad y que se consideran preneoplásicos. Estas lesiones son la metaplasia intestinal y la displasia epitelial.
Por el contrario el tipo difuso de adenocarcinoma gástrico no parece seguir este proceso multipaso de desarrollo y aparentemente se inicia desde una mucosa gástrica sana sin cambios tisulares previos (34).
4. LESIONES PREMALIGNAS
Se definen como aquellos cambios tisulares que pueden progresar a la malignidad y que se encuentran implicados en la carcinogénesis gástrica.
Metaplasia intestinal. Definida como presencia de epitelio diferenciado similar al del intestino delgado. Se clasifica según características morfológicas e histoquímicas en:
-Metaplasia intestinal completa (tipo I).
-Metaplasia intestinal incompleta de intestino delgado (tipo II) o colónica (tipo III).
Así, en la metaplasia intestinal tipo I y tipo II las células caliciformes producen sialomucinas, en tanto que en la de tipo III estas células producen sulfomucinas (55, 56). Aunque parece existir una asociación entre la presencia de metaplasia intestinal y el desarrollo de cáncer gástrico, esta lesión premaligna tiene poca importancia predictiva. La metaplasia intestinal tipo I se relaciona con baja incidencia de presentación de cáncer gástrico en tanto que la metaplasia tipo III tiene un riesgo de 2,7 a 5,8 mayor de desarrollar un cáncer gástrico (57,58).
Displasia epitelial. Caracterizada por la presencia de una serie de alteraciones histológicas como son la atipia celular con pleomorfismo, aumento de células indiferenciadas y disposición anómala de criptas y glándulas.
La displasia epitelial gástrica generalmente ocurre en el contexto de una gastritis crónica atrófica y suele acompañarse de metaplasia intestinal. Con frecuencia se encuentran áreas de displasia alrededor de los adenocarcinomas gástricos y por lo tanto parece clínica y patológicamente relacionada con el cáncer gástrico. Mientras que en la mayoría de los casos, la displasia leve o moderada tiende a regresar o a permanecer estable, la displasia moderada y fundamentalmente la grave están frecuentemente asociadas con el desarrollo de adenocarcinoma gástrico (59) (Fig. 4).
De forma global, en un 10% de los pacientes la displasia epitelial puede progresar a cáncer gástrico en el curso de 5 a 15 años, pero en la mayoría de los pacientes esta displasia regresa o permanece estable (60).
5. CONCLUSIÓN
La carcinogénesis gástrica en un proceso complejo y lento en el que parecen intervenir múltiples factores tanto ambientales como moleculares. Las diferencias geográficas existentes en cuanto a la incidencia, evolución y pronóstico del adenocarcinoma gástrico parecen en parte relacionadas con los diferentes y particulares factores ambientales a los que está expuesta la población (alimenticios e infecciosos). Debido a su más que probable implicación, estos factores han sido los más frecuentemente analizados con resultados concluyentes en relación al acceso y consumo de determinados alimentos, su conservación y su preparación. La infección por H. pylori en la población es otro de los factores implicados en el desarrollo del cáncer gástrico siendo considerado en la actualidad como carcinógeno del grupo I por la OMS.
En cuanto a los factores moleculares, están cobrando cada vez mayor importancia en la carcinogénesis gástrica, jugando un importante papel en el desarrollo de esta enfermedad. La acumulación de alteraciones moleculares parece influir en el inicio y progresión del adenocarcinoma gástrico aunque todavía no está del todo bien establecida su secuencia carcinogénica. Lo que parece claro es que esta secuencia varía según el tipo histológico del adenocarcinoma gástrico. Por este motivo, actualmente se considera que la clasificación del adenocarcinoma gástrico según Lauren, no sólo refleja diferencias histológicas, sino también epidemiológicas, clínicas y pronósticas implicando la posibilidad de la existencia de dos vías de carcinogénesis diferentes que están todavía por establecer.
BIBLIOGRAFÍA
1. Roukos DH. Relevant prognostic factors in gastric Cancer. Ann Surg 2000; 232: 719-20. [ Links ]
2. DeVita VT Jr. The war on cancer has a birthday, and a present. J Clin Oncol 1997; 15: 867-9. [ Links ]
3. Forman D. Are nitrates a significant risk factor in human cancer? Cancer Surv 1989; 8: 443-58. [ Links ]
4. Correa P. Human gastric carcinogenesis: a multistep and multifactorial process-First American Cancer Society award lecture on cancer epidemiology and prevention. Cancer Res 1992; 52: 6735-40. [ Links ]
5. Kramer BS, Johnson KA. Other gastrointestinal cancers: stomach, liver. In: Greenwald P, Kramer BS, Weed DL, eds. Cancer prevention and control. New York: Marcel Dekker, 1995. p. 673-94. [ Links ]
6. Kabat G, Ng S, Wynder E. Tobacco, alcohol intake and diet in relation to adenocarcinoma of the esophagus and gastric cardia. Cancer Causes Control 1993; 4: 123. [ Links ]
7. Howson CP, Hiyama T, Wynder EL. The decline in gastric cancer: epidemiology of an unplanned triumph. Epidemiol Rev 1986; 8: 1-27. [ Links ]
8. Coleman M, Babb P, Damiecki P, Honjo S, Jons J, Knerer G. Cancer survival trends in England and Wales, 1971-1995: deprivation and NHS Region. Studies in Medical and Population Subjets nº 61. National statistics, London, 1999. [ Links ]
9. Stadtländer CT, Waterbor JW. Molecular epidemiology, pathogenesis and prevention of gastric Cancer. Carcinogenesis 1999; 20: 2195-208. [ Links ]
10. Hemminki K, Jiang Y. Familial and second esophageal cancers: a nation-wide epidemiologic study from Sweden. Int J Cancer 2002; 98: 106-9. [ Links ]
11. Kelley JR, Duggan JM. Gastric cancer epidemiology and risk factors. J Clin Epidemiol 2003; 56: 1-9. [ Links ]
12. Mowat C, Carswell A, Wirz A, McColl KE. Omeprazole and dietary nitrate independently affect levels of vitamin C and nitrite in gastric juice. Gastroenterology. 1999; 116: 813-22. [ Links ]
13. Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1984; 16: 1311-4. [ Links ]
14. Bodger K, Crabtree JE. Helicobacter pylori and gastric inflammation. Br Med Bull 1998; 54: 139-50. [ Links ]
15. Forman D. Helicobacter pylori infection and Cancer. Br Med Bull 1998; 54: 71-8. [ Links ]
16. IARC Monogr Eval Carcinog Risks Hum. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon 1994; 61: 1-241. [ Links ]
17. Farthing MJ. Helicobacter pylori infection: an overview. Br Med Bull 1998; 54: 1-6. [ Links ]
18. Cover TL, Blaser M J. Helicobacter pylori: a bacterial cause of gastritis, peptic ulcer disease, and gastric Cancer. Am Soc Microbiol News 1995; 61: 21-6. [ Links ]
19. Asghar RJ, Parsonnet J. Helicobacter pylori and risk for gastric adenocarcinoma. Semin Gastrointest Dis 2001; 12: 203-8. [ Links ]
20. Reed PI, Hill MJ, Johnston BJ. Gastric cancer and Helicobacter pylori. Lancet 1993; 16: 987-8. [ Links ]
21. Kikuchi S. Epidemiology of Helicobacter pylori and gastric cancer. Gastric Cancer 2002; 5: 6-15. [ Links ]
22. Fontana V, Decensi A, Orengo MA, Parodi S, Torrisi R, Puntoni R. Socioeconomic status and survival of gastric cancer patients. Eur J Cancer 1998; 34: 537-42. [ Links ]
23. Solcia E, Fiocca R, Luinetti O, Villani L, Padovan L, Calistri D, et al. Intestinal and diffuse gastric cancers arise in a different background of Helicobacter pylori gastritis through different gene involvement. Am J Surg Pathol 1996; 20 (Supl. 1): S8-22. [ Links ]
24. Watanabe S, Tsugane S, Yamaguchi N. Etiology. In: Sugimura T, Sasako M, eds. Gastric Cancer Oxford University Press, 1997. p. 33-51. [ Links ]
25. Tokunaga M, Land CE, Uemura Y, Tokudome T, Tanaka S, Sato E. Epstein-Barr virus in gastric carcinoma. Am J Pathol 1993; 143: 1250-4. [ Links ]
26. Fukayama M, Hayashi Y, Iwasaki Y, Chong J, Ooba T, Takizawa T, et al. Epstein-Barr virus-associated gastric carcinoma and Epstein-Barr virus infection of the stomach. Lab Invest 1994; 71: 73-81. [ Links ]
27. Shibata D, Weiss LM. Epstein-Barr virus-associated gastric adenocarcinoma. Am J Pathol 1992; 140: 769-74. [ Links ]
28. Ojima H, Fukuda T, Nakajima T, Nagamachi Y. Infrequent overexpression of p53 protein in Epstein-Barr virus-associated gastric carcinomas (abstract). Jpn J Cancer Res 1997; 88: 262-6. [ Links ]
29. Nomura A, Yamakawa H, Ishidate T, Kamiyama S, Masuda H, Stemmermann GN, et al. Intestinal metaplasia in Japan: association with diet. J Natl Cancer Inst 1982; 68: 401-5. [ Links ]
30. Kato I, Tominaga S, Ito Y, Kobayashi S, Yoshii Y, Matsuura A, et al. Atrophic gastritis and stomach cancer risk: cross-sectional analyses. Jpn J Cancer Res 1992; 83: 1041-6. [ Links ]
31. Gordon L. Tumors of the Stomach. En: Sleisenger, Fordtran's, eds. Gastrointestinal and Liver Disease. Philadelphia: Saunders. 2002. p. 733-49. [ Links ]
32. Correa P, Chen VW. Gastric cancer. Cancer Surv 1994; 19-20: 55-76. [ Links ]
33. Hsing AW, Hansson LE, McLaughlin JK, et al. Pernicious anemia and subsequent cancer: a population based cohort study. Cancer 1993; 71: 745-50. [ Links ]
34. Werner M, Becker KF, Keller G, Hofler H. Gastric adenocarcinoma: pathomorphology and molecular pathology. J Cancer Res Clin Oncol 2001; 127: 207-16. [ Links ]
35. Hsu CT, Ito M, Kawase Y, Sekine I, Ohmagari T, Hashimoto S. Early gastric cancer arising from localized Menetrier's disease (abstract). Gastroenterol Jpn 1991; 26: 213-7. [ Links ]
36. Stolte M. Clinical consequences of the endoscopic diagnosis of gastric polyps. Endoscopy 1995 ; 27: 32-7. [ Links ]
37. Parsonnet J, Friedman GD, Vandersteen DP, Change Y, Vogelman JH, Orentreich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991; 325: 1127-31. [ Links ]
38. Blot VJ, Devesa SS, Kneller RW. Rising incidence of adenocarcinoma of the esophagus and gastric cardia. JAMA 1991: 265; 1287-9. [ Links ]
39. Clark GW, Smyrk TC, Buriles P. Is Barrett's metaplasia the source of adenocarcinomas of the cardia? Arch Surg 1994; 129: 609-14. [ Links ]
40. Tahara E. Molecular biology of gastric Cancer. World J Surg 1995; 19: 484-90. [ Links ]
41. Fuchs CS, Mayer RJ. Gastric carcinoma. N Engl J Med 1995; 333: 32-41. [ Links ]
42. Yasui W, Oue N, Kuniyasu H, Ito R, Tahara E, Yokozaki H. Molecular diagnosis of gastric cancer: present and future. Gastric Cancer 2001; 4: 113-21. [ Links ]
43. Ochia, A, Hirohashi S. Multiple genetic alterations in gastric Cancer In: Sugimura T, Sasako M, eds. Gastric Cancer Oxford University Press. 1997. p. 87-99. [ Links ]
44. Wright PA, Williams GT. Molecular biology and gastric carcinoma. Gut 1993; 34: 145-7. [ Links ]
45. Tahara E. Molecular mechanism of stomach carcinogenesis. J Cancer Res Clin Oncol 1993; 119: 265-72. [ Links ]
46. Correa P, Shiao Y H. Phenotypic and genotypic events in gastric carcinogenesis. Cancer Res 1994; 54 (Supl. 7): 1941s-3s. [ Links ]
47. Tahara E, Semba S, Tahara H. Molecular biological observations in gastric Cancer. Semin Oncol 1996; 23: 307-35. [ Links ]
48. Solcia E, Fiocca R, Luinetti O, Villani L, Padovan L, Calistri D, et al. Intestinal and diffuse gastric cancers arise in a different background of Helicobacter pylori gastritis through different gene involvement. Am J Surg Pathol 1996; 20: S8-22. [ Links ]
49. Cho J-H, Noguchi M, Ochiai A, Hirohashi. Loss of heterozygosity of multiple tumor suppressor genes in human gastric cancers by polymerase chain reaction. Laboratory Investigation 1996; 74: 835-41. [ Links ]
50. Uchino S, Tsuda H, Noguchi M, Yokota J, Terada M, Saito T, et al. Frequent loss of heterozygosity at the DCC locus in gastric Cancer. Cancer Res 1992; 52: 3099-102. [ Links ]
51. Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol 1995; 19 (Supl. 1): S37-43. [ Links ]
52. Baffa R, Veronese ML, Santoro R, Mandes B, Palazzo JP, Rugge M, et al. Loss of FHIT expression in gastric carcinoma. Cancer Res 1998; 58: 4708-14. [ Links ]
53. Lauren P. The two histological main types of gastric carcinoma; diffuse and so-called intestinal-type carcinoma. Acta Pathol Microbiol Scand 1965; 64: 31-49. [ Links ]
54. O'Connor F, Buckley M, O'Morain C. Helicobacter pylori: the cancer link. J R Soc Med 1996; 88: 674-8. [ Links ]
55. Filipe MI. Natural history of precursor lesions to gastric carcinoma: growth factors and oncogenes in the metaplasia-dysplasia-carcinoma sequence. Eur J Cancer Prev 1994; 3 (Supl. 2): 19-23. [ Links ]
56. Stemmermann G, Heffelfinger SC, Noffsinger A, Hui YZ, Miller MA, Fenoglio-Preiser CM. The molecular biology of esophageal and gastric cancer and their precursors: oncogenes, tumor suppressor genes, and growth factors. Human Pathol 1994; 25: 968-81. [ Links ]
57. Rokkas T, Filipe MI, Sladen GE. Detection of an increased incidence of early gastric cancer in patients with intestinal metaplasia type III who are closely followed up. Gut 1991; 32: 1110-3. [ Links ]
58. Werner M, Becker KF, Keller G, Hofler H. Gastric adenocarcinoma: pathomorphology and molecular pathology. J Cancer Res Clin Oncol 2001; 127: 207-16. [ Links ]
59. Rugge M, Leandro G, Farinati F, Di Mario F, Sonego F, Cassaro M, et al. Gastric epithelial dysplasia. How clinicopathologic background relates to management. Cancer 1995; 76: 76-82. [ Links ]
60. Rugge M, Farinati F, Baffa R, Sonego F, Di Mario F, Leandro G, et al. Gastric epithelial dysplasia in the natural history of gastric cancer: a multicenter prospective follow-up study. Interdisciplinary Group on Gastric Epithelial Dysplasia. Gastroenterology 1994; 107: 1288-96. [ Links ]

