










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.96 no.9 Madrid sep. 2004
| POINT OF VIEW |
Non-alcoholic steatohepatitis: physiopathological, clinical and therapeutic implications
F. Pérez-Aguilar, S. Benlloch, M. Berenguer, B. Beltrán and J. Berenguer
Service of Digestive Medicine. Hospital Universitario La Fe. Valencia. Spain
Pérez-Aguilar F, Benlloch S, Berenguer M, Beltrán B, Berenguer J. Non-alcoholic steatohepatitis: physiopathological, clinical and therapeutic implications. Rev Esp Enferm Dig 2003; 96: 628-648.
Recibido: 19-02-04.
Aceptado: 18-05-04.
Correspondencia: F. Pérez-Aguilar. Servicio de Medicina Digestiva. Hospital Universitario La Fe. Avda. Campanar, 21. 46009 Valencia. e-mail: fperezaguilar@terra.es
DEFINITION AND PREVALENCE
Non-alcoholic steatohepatitis (NASH) must be considered as a part of a broader-spectrum condition -non-alcoholic fatty liver disease (NAFLD)- that ranges from hepatic steatosis, as its initial form, to NASH, which can progress to liver cirrhosis at the other end of the spectrum. It is usually classified as cryptogenic because of loss of specific characteristics. Histologically, NASH is similar to alcoholic hepatitis and is characterized by macrovesicular steatosis, necroinflammation, hepatocyte ballooning degeneration, and fibrosis. NASH is a chronic disease that is very frequently detected in patients with impaired liver function. Most patients are female, obese with type-2 diabetes mellitus and/or hyperlipidemia with insulin resistance. However, it may also appear in lean males and females with no associated pathology, and in relation to a variety of situations such as diversion surgery, medication, etc. Powell et al. (1) defined NASH as the presence of characteristic histological findings with persistently elevated transaminase levels, no previous history of significant alcohol intake (< 30 g a week corroborated by family) and no other type of hepatic disease.
Since it was first described by Ludwig in 1980 (2), the epidemiological impact and the number of recent publications on this condition have increased. The prevalence of NAFLD in the general population is estimated to be 20%, and 2-3% for NASH, which makes NASH the potentially most common hepatic disease in the United States (3-5). NAFLD has a higher prevalence in patients with persistently elevated transaminase levels, and is estimated to range from 21 to 63% (6). In morbidly obese patients requiring gastric diversion surgery, NAFLD has a prevalence of 95 versus 25% for NASH (7). NAFLD occurs in 63% of patients with type-2 diabetes mellitus (8). It must be emphasized that obesity is increasing in the American population, and may become as high as 40% by 2025 (9). Consequently, the prevalence of NAFLD, including NASH, will notably increase.
NATURAL HISTORY
Information is scarce on the natural history of this disease, which can progress to the following consecutive stages in some patients: fatty liver, steatohepatitis, steatohepatitis with fibrosis, and cirrhosis.
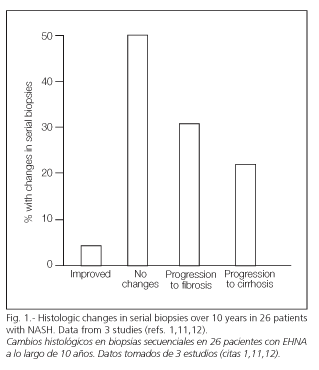
Matteoni et al. (10) reported that of 132 patients with NAFLD, 25% had already developed NASH in progression to cirrhosis at 10 years' follow-up, with 12% dying due to liver disease. Other studies (4,11-13) have corroborated the possible progression of this condition in a significant percentage of patients (Fig. 1). Similarly, many patients with cryptogenic cirrhosis may develop NASH with a loss of the peculiar histological characteristics of this condition (14,15). These cryptogenic cirrhosis may even recur in the form of NASH following transplantation (16).
Factors that may imply a higher risk of steatosis developing to NASH include: age over 40 years, body mass index > 40 kg/m2, AST/ALT ratio > 1, and the presence of at least two causes of NASH, such as diabetes and hyperlipemia (17,18).
Many factors can contribute to NASH-related mortality, including the complications of obesity and diabetes. Causes of liver disease-related mortality include liver failure, cirrhosis complications (hemorrhage due to varices or ascites), and hepatocarcinoma, although the precise incidence of each of these complications is unknown (19).
Histological improvement can also occur, especially in those with minimal fibrosis. Following weight loss, a drop in inflammation and Mallory bodies may be detected -including perisinusoidal fibrosis- particularly if weight is gradually lost and diet is associated with physical exercise (20,21). In many cases liver failure manifests during rapid weight loss, regardless of the method used, especially in patients with morbid obesity undergoing weight-loss surgery (22,23).
MAJOR CONDITIONS ASSOCIATED WITH NASH
Insulin resistance plays a fundamental role in type-2 diabetes mellitus, as well as in obesity, and is the most predisposing and reproducible factor in NASH (24) (Table I).

Diabetes mellitus
Up to one third of patients have diabetes or fasting hyperglycemia at the time of diagnosis with NASH (12,25). The most frequent association is type-2 diabetes, although difficult-to-control insulin-dependent diabetes may also be present (26). Diabetes is an important independent predictor of severe hepatic fibrosis in NASH (17). Furthermore, glucose intolerance occurs secondarily to cirrhosis in 60-80% of these patients, 10-30% of whom eventually develop diabetes (27). Autopsy studies have revealed that type-2 diabetes is associated with NASH, with a 2.6-fold increased risk (28).
Obesity
Between 39 and 100% of patients with NASH are overweight (BMI > 25 kg/m2) or obese (> 30 in Caucasians and > 27 in Asians) (25, 29-31). An autopsy study found the prevalence of NASH to be 6-fold greater among obese versus lean individuals (28). Liver biopsy demonstrates steatosis in more than 75% of the morbidly obese; NASH is identified in a quarter of these patients, and cirrhosis in 3-11% (32). Obesity also correlates with the severity of fibrosis in NASH, regardless of the diabetes status or age (18). García-Monzón et al. (33) found that the older the age and the higher the grade of steatosis, particularly if intrahepatic inflammation is also present, the higher the risk of fibrosis.
Distribution of body fat may be important; a significant correlation has been found between grade of steatosis and waist-to-hip ratio (33), indicating the importance of visceral fat as a predictor of steatosis (34). Lean males rarely present with significant hepatic fibrosis; Ratziu et al. (18) did not find fibrosis or cirrhosis in patients younger than 50 years with a BMI < 28 kg/m2 and normal triglyceride values.
Hyperlipemia
Between 20 and 80% of patients with NASH have hyperlipemia (25). Although triglyceride and/or total serum cholesterol levels are increased, there is a greater association with triglyceride values, especially for hyperlipemias 2b and 4, versus 2a (35). A relevant role has been ascribed to hypertriglyceridemia in the pathogenesis of NASH, since a correction of this lipid anomaly has been associated with improved liver function tests (36).
Rapid weight loss
NASH has been associated with rapid weight loss following surgery for obesity, extensive intestinal resection, and severe fasting (37).
Age and sex
Older age and female sex are considered independent factors associated with liver fibrosis in NASH (17,37).
Other conditions associated with NASH
Jejunoileal bypass, gastroplasty, gastric bypass and other surgical techniques leading to rapid weight loss, as well as feeding disorders such as anorexia or bulimia, celiac disease, jejunal diverticulosis, other causes of bacterial overgrowth, total parenteral nutrition, abetalipoproteinemia, partial lipodystrophy, Weber-Christian disease, toxic oil syndrome, etc., are related to NASH development (37).
Drug-induced NASH
Cardiovascular drugs such as amiodarone, perhexiline maleate, and more rarely calcium channel blockers such as nifedipine and diltiazem, high-dose glucocorticoids, synthetic estrogens, tamoxifen, chloroquine, etc., may also be associated with NASH (37).
Amiodarone, perhexiline maleate, diethylaminoetoxyhexestrol and tamoxifen cross the mitochondrial external membrane without difficulty because of their lipophilic properties, and are "pushed' into the mitochondria from the intermembranous space by the high electrochemical potential at the internal membrane, thus reaching high intramitochondrial levels. Beta-oxidation (causing steatosis) is thus inhibited, and electron transfer throughout the respiratory chain is blocked, which leads to a transfer of electrons to oxygen, thereby forming superoxide radical anions and generating oxygen free radicals.
ETIOPATHOGENESIS OF NASH
Insulin resistance appears to be a key pathogenic and reproducible factor in NASH (30,39,40). It is defined as a reduced capacity of insulin to perform its biological functions in typical target tissues such as musculoskeletal, liver or fat tissues. This concept falls within the so-called metabolic syndrome or syndrome X in which several clinical diseases are associated, including obesity, hyperlipemia, arterial hypertension, and diabetes mellitus, and which carries a higher risk for cardiovascular disease. Hyperinsulinemia basically results from compensatory hypersecretion by beta-cells and not, as previously believed, from reduced insulin breakdown as a result of liver disease, although this mechanism may also have an influence in cirrhotics.
Insulin-stimulated intracellular glucose transport is preferably controlled by a translocation of the GLUT4 carrier from an intracellular vesicular membrane to the plasma membrane, which occurs after the binding of insulin to its cell receptor. GLUT4 expression is altered in different forms of insulin resistance; e. g., a reduced expression of the gene GLUT4 has been found in fat cells of obese individuals, diabetics, or those with glucose intolerance, without altering the degree of expression in other cells such as musculoskeletal cells (41).
A hypothesis has recently been put forward that fat cells may play a central role in the development of insulin resistance, as well as of NASH. Fat cells appear to be an important endocrine organ that may trigger an inflammatory process in relation to NASH development. It can secrete potentially toxic substances such as tumor necrosis factor (TNF), leptin, resistin, and fatty acids whose concentration levels correlate with insulin resistance, and therefore they should be relevant in the development of type-2 diabetes. Furthermore, it has been observed that visceral fat (and not total body fat) is a predictive factor of liver steatosis, hyperinsulinemia, reduced hepatic extraction of glucose, and insulin resistance.
For a better understanding of the emerging hypotheses on NASH, it may be appropriate to review the normal metabolism of fatty acids. During digestion, dietary triglycerides are converted by enterocytes into chylomicrons, which then migrate via the lymphatics and are subsequently hydrolyzed into fatty acids by lipoprotein lipase at the capillary endothelium of adipose and liver tissues. Free fatty acids thus produced are highly miscible with cell membranes, so that they immediately go to fat cells where they are converted into resterified triglycerides, or to the muscle as energy supply, or enter the liver. During fasting, fatty acids supplied to the liver result from a hydrolysis of triglycerides stored within the adipose tissue. Thus, under normal conditions hydrolysis is stimulated by catecholamines, glucagon, and growth hormone, and is inhibited by insulin. In the liver, fatty acids undergo mitochondrial beta-oxidation, a step of triglyceride synthesis or phospholipid and cholesterol ester formation.
However, in situations of insulin resistance, as is the case with most patients with NASH, increased blood levels affect the adipocyte and liver cells in a different way. In the adipocyte it favors lipolysis with the consequent release of more fatty acids to the liver; in the hepatocyte it stimulates fatty acid synthesis and inhibits mitochondrial beta-oxidation of fatty acids (42). Furthermore, high levels of insulin can inhibit apolipoprotein B100, a component of VLDL, synthesis, which makes it difficult for triglycerides to be transported out of the liver.
The greater afflux of fatty acids to the liver, together with the potential alterations of its metabolization within the liver (including greater triglyceride synthesis, reduced triglyceride elimination, and reduced beta-oxidation of fatty acids), results in hepatic steatosis; these mechanisms are considered a "first impact' in the development of NASH. However, steatosis is not always quiescent, since high intrahepatic concentrations of free fatty acids and their saturation of mitochondrial beta-oxidation make them susceptible to a "second impact', where additional factors influencing oxidative stress and lipid peroxidation are involved; this leads to a high afflux of electrons to the mitochondrial respiratory chain, and an increased production of oxygen free radicals (OFR), which are responsible for the hepatic lesions of NASH (Fig. 2).

Furthermore, under conditions of high fatty acid metabolism, OFR can also result from peroxisomal beta-oxidation and microsomal omega-oxidation by cytochrome P450 (CYP2E1, CYP4A) of fatty acids (43,44). Patients with NASH have been shown to present ultrastructural mitochondrial lesions (40,45), reduced activity of mitochondrial complexes (46), and deficient mitochondrial ATP formation (47), which are also involved in OFR formation.
OFR determine the production of various cytokines in different types of cells (hepatocytes, adipocytes, and Kupffer cells). The foregoing depends on the activation capacity of OFR on transcription factors, basically the nuclear factor κB (NF-κB). This factor is normally found synthesized and maintained in an inactive form within the cell cytoplasm, bound to the IKK protein. OFR also have the capacity, at appropriate concentration levels, to activate the IKK-ß enzyme, which unbinds NF-κB from IKK, rendering the factor free to migrate to the nucleus and to start transcription processes. These processes determine the formation of cytokines such as tumor necrosis factor-alpha (TNF-α), tumor growth factor-beta 1 (TGF-ß1), Fas ligands, and interleukin-8, among others. In turn, each one of these cytokines may be involved in the pathogenesis of hepatic lesions. TNF-aα and TGF-ß1 can activate programmed cell death or apoptosis when caspases -the enzymes responsible for the foregoing- are activated. TGF-ß1 can intervene in the formation of Mallory bodies and activate collagen synthesis by stellate cells, and IL-8 is a potent neutrophil activator (44,47) (Fig. 3).

It should be pointed out that the products derived from lipid peroxidation, melandialdehyde (MDA) and 4-hydroxynonenal (HNE) (48), appear to be involved in the pathogenesis of NASH-related hepatic lesions. Both cause direct toxicity and can trigger immune reactions when they covalently bind to proteins (Fig. 3); they may induce the formation of Mallory bodies by promoting a clustering of cytokeratins, and can also increase collagen synthesis by activating hepatic stellate cells. HNE also has a chemotactic activity on neutrophils (Fig. 3)
Once NF-κB is activated, hepatocytes can synthesize Fas ligand, and then secrete it; this in turn may act on the Fas receptors of adjacent hepatocytes, thereby producing fratricidal killing among these (49,50).
Other factors involved in the pathogenesis of NASH
Most patients with primary iron overload unrelated to hemochromatosis have insulin resistance (51-54), which may improve with phlebotomy (55,56). Insulin resistance causes a greater expression of transferrin receptors on the cell surface, and increases the exocytosis of pre-existing intracellular receptors in association with high concentrations of serum ferritin (12,51,53) and increased liver iron in some patients (57). Elevated ferritin does not necessarily mean increased liver iron, but is due to NASH as an acute phase reactant. Ferrous iron is a potent generator of hydroxyl radicals and can contribute to OFR accumulation, cell injury, and cell death; when stellate cells are activated, it can stimulate fibrogenesis. It has yet to be determined whether moderate iron overload in NASH participates in the pathogenesis of this disease, or is related to associated metabolic anomalies, or is due to unidentified environmental or genetic factors. For example, heterozygous mutation of the HFE gene, frequently detected in these patients, might increase iron deposition in the liver (58).
It has been suggested that leptin may be classified as a cytokine as it does not only regulate food intake and energy consumption, but also modulates immune and inflammatory responses (59). High serum levels of leptin appear to correlate with the severity of hepatic steatosis, which suggests a pathogenic role of leptin in hepatic insulin resistance and/or a failure of its anti-steatotic activity. Furthermore, its production by stellate cells may play an important role in hepatic fibrosis (60-62). It can contribute to the progression of steatosis to NASH and finally to cirrhosis, given its profibrogenic and modulating activity on the hepatic inflammatory response (63). On the other hand, it has been observed that the administration of leptin to congenitally leptin-deficient mice with generalized lipodystrophy induces a reduction in body fat and a marked reduction of insulin resistance (64).
Endotoxin and endotoxin-mediated cytokine release plays an important role in the pathogenesis of alcoholic hepatitis. In the foregoing situation, there is an increased production of TNF-α, IL-6, and IL-8 that favors necrosis and the inflammatory response. Endotoxin can also contribute to the development of NASH in some cases, as in those arising from intestinal diversion surgery. Experimentally, in genetically obese rats there is a significantly increased production of endogenous ethanol, an enhanced sensitivity to endotoxin, and an alteration of Kuppfer cells, all of which favor the development of NASH. Furthermore, it has been observed that the production of endogenous ethanol decreases after treatment with oral neomycin; therefore one could hypothesize on whether the small amount of ethanol detected in some patients as produced by the intestinal flora could be involved in the pathogenesis of this disease (65).
Proliferating peroxisome activated receptors (PPAR) are expressed in tissues with important oxidative phosphorilation, and regulate lipids through the peroxisomal, microsomal, and mitochondrial pathways. Some mutations of the encoding gene for these nuclear receptors have been identified in patients with NASH, and might be involved in its pathogenesis (66).
The possible role of different drugs in the pathogenesis of this disease is discussed in a previous section.
DIAGNOSIS
Although most cases of NASH are detected in the fifth and sixth decades of life, it should be emphasized that the prevalence of this disease is increasing in children (67,68); it can therefore present at any age.
Clinical manifestations and laboratory anomalies
Most patients (45-50%) are asymptomatic, but a small percentage (10), especially children (67,68), may present symptoms such as pain in the right hypochondrioum, abdominal discomfort, asthenia, and malaise (69). They typically suffer from other diseases, and anomalies of liver function or hepatomegaly are incidentally discovered (10); the latter condition has been detected in 12-75% of cases (10,17,70-72). It is interesting to remember that many cases of cryptogenic cirrhosis could be the end stage of NASH and present with the multiple complications of advanced cirrhosis.
Several studies (73,74) suggest that hepatocarcinoma could be a complication of this disease. In genetically obese ob/ob leptin-deficient mice, that rarely develop cirrhosis, an increased incidence of hepatocarcinoma has been observed, an incidence similar to that found in obese and diabetics subjects. According to this study, metabolic anomalies might facilitate the progression of NASH to hepatocarcinoma (75). In patients with cryptogenic cirrhosis, the incidence of hepatocarcinoma was found to be higher than in cirrhotic patients with a well-defined viral or alcoholic etiology (76), and therefore hepatocarcinoma could be a late complication of NASH. However, more recent studies have shown an association of diabetes with hepatocarcinoma only in the presence of hepatitis C virus, hepatitis B virus, or alcoholic cirrhosis (77), which suggests that diabetes may only be a marker of advanced liver disease with a greater likelihood of progression to hepatocarcinoma.
The most frequent anomaly in liver function tests in this disease is a 2-5-fold increase in transaminases (10, 17,71), occasionally a 10-15-fold increase (17), although they normally remain within normal values. A differential feature versus alcoholic liver disease is that the AST/ALT ratio is usually below 1 in 65-90% of patients with NASH (10,20,64,67). An AST/ALT ratio greater than 1 suggests an advanced form of NASH (10,20). Alkaline phosphatase and gamma-glutamiltranspeptidase may be 2 or 3-fold higher in more than 50% of patients (10, 69,70,72). Bilirubin and albumin usually remain within their normal ranges (7,69,70,72).
George et al. (78) and Bonkovsky et al. (58) reported that patients with NASH and iron overload had a greater hepatic compromise and more acute fibrosis, which they ascribed to a greater prevalence of the C2/2Y mutation. Moirand et al. (79) described a new syndrome of iron overload in relation to metabolic disorders and insulin resistance. Younossi et al. (80) and Angulo et al. (17) did not find any relationship between iron and clinical or histological manifestations in patients with NASH. Fargion et al. (81) observed that patients with hepatic steatosis often presented with elevated ferritin levels and normal transferrin saturation, with iron overload in those in which it persists despite an appropriate diet; the simultaneous disorder of iron, glucose and/or lipid metabolism associated with insulin resistance in most cases is responsible for persistent hyperferritinemia, and may help identify those patients at risk for NASH. Mendler et al. (57) did not detect a greater iron overload in patients with NASH versus those with hepatic steatosis, and the HFE gene did not influence hepatic injury, although an un-explained hepatic iron overload almost consistently appears in association with the insulin resistance syndrome. Chitturi et al. (82) found that patients with NASH often had hyperferritinemia but with normal transferrin saturation, and HFE mutations do not confer a greater risk of fibrosis.
Hepatic steatosis can cause primary graft dysfunction following liver transplantation; therefore livers with severe steatosis (more than 60% of hepatocytes with fatty vacuoles) should be completely discarded for transplantation, and it should be taken into account that grafts with moderate steatosis (30-60% of hepatocytes with fatty vacuoles) carry a risk.
Exclusion of other diseases
Other diseases of the liver can be associated with NAFLD, and the latter can influence the prognosis of conditions such as hepatitis C virus-related cirrhosis or HFE-hemochromatosis; therefore, tests positive for the C virus or hemochromatosis do not exclude the diagnosis of NAFLD. Steatosis can also occur in Wilson's disease, autoimmune liver disease, galactosemia, and alcoholic liver disease. Thus, pertinent studies should be performed to rule out other chronic hepatic diseases such as hepatic disease caused by the B or C virus, primary biliary cirrhosis, primary sclerosing cholangitis, hemochromatosis, porphyria, and those of toxic origin.
Liver biopsy
There is a poor correlation between clinical, laboratory, and pathological findings in NAFLD; therefore, it is impossible to stage patients with NAFLD without a histological study. Biopsy also permits a determination of liver iron concentration, which some authors have observed to be increased in relation to the C282Y mutation, and which increases the risk of fibrosis in a group of patients with NASH. Because of all the foregoing reasons, many hepatologists advocate that a liver biopsy be performed in all patients with a presumptive diagnosis of NAFLD (in spite of the risk of this procedure and the limited treatment options currently available). Furthermore, it can provide more insight into the natural history of the disease and evaluate the influence of different treatments. On the other hand, although hepatic steatosis generally carries a benign prognosis and can be diagnosed by clinical, laboratory, and ultrasound evaluation, it can progress to NASH and cirrhosis; therefore, patients whose data are suggestive of disease progression should be selected and entered into treatment protocols. Although histological assessment is the golden rule for identifying NASH, there is no widespread agreement in regard to its evaluation, although Brunt modified semiquantitative staging (83), which classifies inflammatory activity into grades 0 to 3 and fibrosis into stages 0 to 4, is the most widely used system (Table II). Lesions are similar, but not identical, to those of alcoholic steatohepa-titis, including generally macrovesicular steatosis, ballooning degeneration of hepatocytes, mixed acute and chronic, mild, diffuse, lobular inflammation (neutrophils and T lymphocytes), and perivenular and perisinusoidal collagen deposits; these lesions can be more marked in Rappaport zone III; Mallory hyaline, vacuolated periportal hepatocyte nuclei, lobular lipogranuloma, and PAS-diastase-resistant Kuppfer cells are common findings. Portal inflammation can be more evident in children than in adults. The progression of fibrosis can result in bridging septae formation and cirrhosis. NASH can occur concurrently with other hepatic diseases. Lesions necessary for the diagnosis of NASH, as well as other less frequent and even unlikely lesions, are listed in table III.


Ultrasound and computed tomography
Ultrasounds have a sensitivity of 89% and a specificity of 93% for the detection of steatosis (84), but they cannot distinguish between the various precirrhotic states of fibrosis, and its maximum value is in the diagnosis of hepatic cirrhosis, for which it has a sensitivity of 45-80% and a specificity of 63-91%; computed tomography has a higher specificity, but is more expensive.
Insulin resistance
It can be studied by:
1. the HOMA method (homeostasis model assessment), applying the formula of Matthews where insulin resistance = fasting serum insulin (µU/ml) x glucose (mmol/L)/22.5, considering insulin resistance as HOMA values > 3.8 (3.9 in females and 3.5 in males) (52,60,85,86).
2. Serial determinations of glucose, insulin and C peptide levels, during frequent performance of intravenous glucose tolerance tests; the latter method allows measurement of insulin secretion and an estimation of hepatic insulin extraction by C peptide determination (60,87,88).
TREATMENT
The fact that there is no universal effective treatment for NASH leads some to avoid invasive diagnostic tests such as liver biopsy. Further insight into the natural history of the disease and prospective therapeutic trials for correct decision-making are warranted (89). Future therapeutic trials for NASH should be randomized, placebo-controlled, double-blind studies including a greater number of patients for longer periods, and they should also assess histological lesion grade both before and after treatment. Combination therapies (to improve the response to insulin and reduce oxidative stress) must be compared with different monotherapy regimens.
Change in habits
Diet and physical exercise significantly reduce the risk of developing type-2 diabetes (90). Given the important relationship between insulin resistance and NAFLD, a change in habits is advisable. Reducing weight can improve hepatic enzyme alterations and histology in patients with NASH (91,92). Dietary restriction and physical exercise led to improved liver enzymes in obese patients who lost weight versus patients whose weight remained unchanged or increased, and serial biopsies also showed a significant improvement in hepatic steatosis (93).
Andersen et al. (23) conducted a study on 41 morbidly obese patients treated with a very low calorie diet (288 kcal/day) that achieved an average weight loss of 34 kg, and found an improvement in the laboratory parameters of most patients; serial biopsies also showed some improvement of steatosis. However, 20% of patients, especially those that had lost weight faster, showed increased portal inflammation and fibrosis, indicating that gradual weight loss is beneficial but rapid weight loss can be harmful. Some experts recommend a gradual weight loss of about 10% within 6 months.
Lipid-lowering drugs
A randomized trial on gemfibrozil (94) 600 mg daily for four weeks showed a significant improvement of transaminases, which did occur in the control group. The administration of clofibrate 2 mg daily for one year showed no changes in analytical findings, steatosis, inflammation, or fibrosis (95). In two patients with breast cancer who received bezafibrate for tamoxifen-induced NASH, subsequent computer tomographic assessments could not detect hepatic steatosis (96). By acting as a ligand of the proliferating-activated peroxysomal α receptor, this drug could compensate for the insufficient mitochondrial ß oxidation in patients with NASH. Treatment with probucol, a lipid-lowering drug with potent antioxidant properties, at a dose of 500 mg/day for 6 months, achieved a significant reduction in transaminase levels in 27 patients with NASH (97). The preliminary results of treatment with atorvastatin are promising and indicate that 3-hydroxy-3methylglutaryl-coenzyme A reductase inhibitors may be useful in the treatment of NASH (98).
Drugs that improve insulin sensitivity
In insulin resistance and fatty liver models in mice, treatment with thiazolidinedione (which acts as a ligand with a strong affinity for activated proliferating peroxysome ω receptors) or metformin improved both conditions (99,100). The administration of troglitazone significantly improved impaired liver function and also caused a certain degree of histological improvement in patients with NAFLD (101). This drug has been withdrawn from the market as a first line therapy for diabetes due to some cases of potentially lethal liver toxicity. A second generation of thiazolidinediones, such as darglitazone, rosiglitazone, or pioglitazone, with a smaller potential for liver toxicity, may be useful in the treatment of NAFLD. Newschwander-Tetri (102) administered rosiglitazone 4 mg/12 h for 48 weeks to 30 patients with NASH. All patients were overweight and half of them had hydrocarbon intolerance or diabetes. Treatment achieved an improvement of the necrotic inflammatory process as seen in liver biopsy in 45%, and transaminase levels returned to almost normal levels at the end of treatment, but later increased to almost pretreatment levels. Treatment with pioglitazone 45 mg daily for 6 weeks (103) in 11 patients with type-2 diabetes increased plasma adiponectin, reduced liver fat, and improved peripheral and hepatic sensitivity to insulin; therefore, it may have an important role in mobilizing hepatic fat for type-2 diabetic patients.
Metformin, a biguanide that reduces hyperinsulinemia and improves hepatic insuline resistance, is used as an oral antidiabetic. In a small group of patients with NAFLD (39), liver enzymes and steatosis also significantly improved, perhaps due to a reduced hepatic expression of TNF-α, a cytokine that promotes insulin resistance and can incite the characteristic necroinflammatory lesions of NASH. There is sufficient evidence that this drug can be used relatively safely in patients with NASH, insulin resistance, and prediabetes (104). It is contraindicated for patients with potential lactic acidosis, renal failure, or congestive heart failure. In a study on the prevention of diabetes, fenformin was well tolerated in a wide group of insulin-resistant obese patients, and significantly reduced the incidence of established diabetes (90).
Leptin is considered to contribute to the appearance of steatosis and NASH by promoting insulin resistance, altering hepatocyte insulin signaling, promoting fatty acid synthesis, and increasing proinflammatory responses; however, it could be beneficial in lipodystrophic patients with NAFLD, since the administration of leptin to lipodystrophic mice that had developed secondary leptin deficiency, associated with insulin resistance and steatosis, led to their healing (64).
Ursodeoxycolic acid
In three pilot studies (95,105,106) this hydrophilic bile acid with membrane-stabilizing, cytoprotective, and immunomodulating properties was found to improve or normalize liver function. However, in another randomized study that included 166 patients with histologically demonstrated NASH under treatment with 13-15 mg/kg/day of ursodesoxycolic acid or placebo for two years no significant changes were found in the degree of steatosis, necroinflammation or fibrosis between both groups at the end of the study (107). Further controlled and randomized trials in larger numbers of patients are warranted, perhaps using higher doses (15-20 mg/kg/day) and for longer periods.
Antioxidants
Oral vitamin E 400-1200 IU daily improves hepatic enzymes in patients with, and also in certain animal models of, NAFLD (108,109). It might improve NASH by modulating cytokines (110), and inhibiting the expression of intrahepatic TGF-ß1, which is involved in fibrogenesis. Because it is well tolerated and low-cost, its use in the treatment of this condition is appealing. Betaine is a choline metabolite that increases S-adenosylmethionine levels, protects the liver from triglyceride deposits and oxidative stress in mice fed with alcohol, and may therefore have some efficacy as an antioxidant in NAFLD (111). In a pilot study carried out in 10 patients with NASH (112), betaine reduced hypertransaminasemia and achieved histological improvement of steatosis and necroinflammation.
Silimarine is derived from herbs commonly used in the treatment of hepatic diseases, and also has antioxidant properties. It stimulates ribosomal polymerase RNA, is an iron chelator, and reduces tumor promotion activity. Velussi (113) evaluated the efficacy of silimarine in reducing lipid peroxidation and insulin resistance in diabetics with alcoholic cirrhosis. At 12 months, baseline glucose levels, insulin levels, and malondialdehyde levels decreased.
N-acetylcysteine increases hepatic gluthatione levels and protects the liver from oxidative stress. Eleven patients with histologically demonstrated NASH who were treated with N-acetylcysteine 1000 mg/day for 3 months showed a significant decrease in transaminase and gamma-glutamyltranspeptidase levels; one patient was withdrawn from the study due to epigastralgia (114).
Antibiotics
In rats receiving total parenteral nutrition the administration of polymixin B and metronidazole reduced the degree of hepatic steatosis and TNF-α production, most of which comes from Kuppfer cells that might have been activated by endotoxins (115,116).
Nutritional supplementation
Carnitine and/or choline deficiency may contribute to the appearance of steatosis or NASH by reducing free fatty acid oxidation and VLDL secretion. In a group of patients receiving prolonged total parenteral nutrition, intravenous choline supplementation returned plasma choline levels to normal, and steatosis completely disappeared, which was confirmed by CT (117).
Therapeutic phlebotomies
They are another option that should be considered for patients with NASH and iron overload, as mentioned earlier (55,56). Iron depletion has been beneficial regarding coronary response, endothelial dysfunction, insulin secretion, insulin action, and metabolic control of type-2 diabetes (118). Guillygomarc'h et al. (119) pointed out that performing a phlebotomy in patients with hepatic iron overload associated with insulin resistance not only achieved a subjective improvement without adverse effects, but can also reduce the risk of hepatic fibrosis, cancer, and cardiovascular disease. Facchini et al. (120) observed that phlebotomy performed in carbohydrate-intolerant patients and NAFLD patients improved plasma insulin levels during fasting and after glucose stimulation, and transaminase returned to nearly normal levels, which shows the insulin-saving effect of iron depletion and the important role of iron and hyperinsulinemia in the pathogenesis of NAFLD.
Liver transplantation
It may be indicated in patients with end-stage hepatic cirrhosis secondary to NASH. Although liver transplantation usually has a good outcome, NASH recurrence may occur after transplantation (121-124). The causes of recurrence are unknown and may probably be due to multiple factors, including hypertriglyceridemia, obesity, diabetes, and corticoid therapy. It must be underscored that severe -even moderate- hepatic steatosis may result in primary graft dysfunction; therefore, grafts with this condition are not acceptable for transplantation.
ACKNOWLEDGEMENTS
This study was supported in part by grant C03/02 of Red Nacional de Centros de Investigación, Instituto de Salud Carlos III.
REFERENCES
1. Powell EE, Cooksley WGE, Hanson R, Searle J, Halluday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology 1990; 11: 74-80. [ Links ]
2. Ludwig J, Viggiano RT, McGill DB. Non-alcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin Proc 1980; 55: 342-8. [ Links ]
3. El-Hassan AY, Ibrahim EM, Al-Mulhim FA, Nabhan AA, Chamas MY. Fatty infiltration of the liver: analysis of prevalence, radiological and clinical features and influence of patients management. Br Radiology 1992; 65: 774-8. [ Links ]
4. Bellentani S, Saccoccio G, Masutti F, Croce LS, Brandi G, Sasso F, et al. Prevalence of and risk factors for hepatic steatosis in northern Italy. Ann Intern Med 2000; 132: 112-7. [ Links ]
5. Jick SS, Stender M, Myers MW. Frequency of liver disease in type 2 diabetic patients treated with oral antidiabetic agents. Diabetes Care 1999; 2: 1067-71. [ Links ]
6. Hulcranzt R, Glaumann H, Lindberg G, Nilsen HL. Liver investigation in 149 asymptomatic patients with moderately elevated activities of serum aminotransferases. Scand J Gastroenterol 1986; 21: 109-13. [ Links ]
7. Dixon JB, Bhathal PS, O´Brien PE. Nonalcoholic fatty liver disease: predictors of nonalcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology 2001; 121: 91-100. [ Links ]
8. Kemmer NM, McKinney KH, Xiao SY, Singh H, Murray R, Abdo B, et al. High prevalence of NASH among Mexican American females with type II diabetes mellitus (Abstract). Gastroenterology 2001; 120: A117. [ Links ]
9. Kopelman PG. Obesity as a medical problem. Nature 2000; 404: 635-43. [ Links ]
10. Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999; 116: 1413-9. [ Links ]
11. Lee RG. Non-alcoholic steatohepatitis: a study of 49 patients. Hum Pathol 1989; 20: 594-8. [ Links ]
12. Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Nonalcoholic steatohepatitis: an expanded clinical entity. Gastroenterology 1994; 107: 1103-9. [ Links ]
13. Hilden M, Christoffersen P, Juhle E, Juhl E, Dalgaard JB. Liver histology in a "normal' population-examination of 503 consecutive fatal traffic casualties. Scand J Gastroenterol 1977; 12: 593-7. [ Links ]
14. Caldwell S, Oelsner D, Iezzoni J, Hespenheide E, Battle E, Driscoll C. Cryptogenic cirrhosis. Clinical characterization and risk factors for underlying disease. Hepatology 1999; 29: 664-9. [ Links ]
15. Poonawala A, Nair S, Thuluvath P. Prevalence of obesity and diabetes in patients with cryptogenic cirrhosis. Hepatology 2000; 32: 689-91. [ Links ]
16. Ong J, Younossi ZM, Reddy V, Price LL, Gramlich T, Mayes J, et al. Cryptogenic cirrhosis and postransplant nonalcoholic fatty liver disease. Liver Transpl 2001; 7: 797-801. [ Links ]
17. Angulo P, Keach JC, Batts KP, Batts KP, Lindor KD. Independent predictors of liver fibrosis in patients with nonalcoholic steatohepatitis. Hepatology 1999; 30: 1356-62. [ Links ]
18. Ratziu V, Giral P, Charlotte F, Bruckert E, Thibault V, Theodoriu I, et al. Liver fibrosis in overweight patients. Gastroenterology 2000; 118: 1117-23. [ Links ]
19. Sanyal AJ. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology 2002; 123: 1705-25. [ Links ]
20. Ueno T, Sugawara H, Sujaku K, Hashimoto O, Tsuji R, Tamaki S, et al. Therapeutic effects of restricted diet and exercise in obese patients with fatty liver. J Hepatol 1997; 27; 103-7. [ Links ]
21. Vajro P, Fontanella A, Perna C, Orso G, Tedesco M, De Vincenzo A. Persistent hyperaminotransferasemia resolving after weight reduction in obese children. J Pediatr 1994; 125; 239-41. [ Links ]
22. Dean RH, Scott HW jr, Shull HJ, Gluck FW. Morbid obesity: problems associated with operative management. Am J Nutr 1977; 30: 90-7. [ Links ]
23. Andersen T, Gluud C, Franzmann MB, Christoffersen P. Hepatic effects of dietary weight loss in morbidily obese subjects. J Hepatol 1991; 12: 224-9. [ Links ]
24. Belfiore F, Iannello S. Insulin resistance in obesity: metabolic mechanisms and measurement methods. Mol Genet Metab 1998; 65: 121-8. [ Links ]
25. James OFW, Day CP. Nonalcoholic steatohepatitis (NASH): a disease of emerging identity and importance. J Hepatol 1998; 29: 495-501. [ Links ]
26. Lenaerts J, Verresen L, Van Steenbergen W, Fevery J. Fatty liver hepatitis and type 5 hyperlipoproteinemia in juvenile diabetes melllitus. Case report and review of the literature. J Clin Gastroenterol 1990; 12: 93-7. [ Links ]
27. Petrides AS, Vogr C, Schulze-Berge D, Schulze-Berge D, Matthews D, Strohmeyer G. Pathogenesis of intolerance and diabetes mellitus in cirrhosis. Hepatology 1994; 19: 614-27. [ Links ]
28. Wanless IR, Lentz JS. Fatty liver hepatitis (steatohepatitis) and obesity: an autopsy study with analysis of risk factors. Hepatology 1990; 12: 1106-10. [ Links ]
29. Andersen T, Gluud C. Liver morphology in morbid obesity: a literature study. Int J Obes 1984; 8: 97-106. [ Links ]
30. Marceau P, Biron S, Hould F-S, Hould FS, Marceau S, Simard S, et al. Liver pathology and the metabolic syndrome X in severe obesity. J Clin Endocrinol Metab 1999; 84: 1513-7. [ Links ]
31. Naveau S, Giraud V, Borotto E, Aubert A, Capron F, Chaput JC. Excess weight risk factor for alcoholic liver disease. Hepatology 1997; 25: 108-11. [ Links ]
32. Luyckx FH, Lefebvre PJ, Scheen AJ. Nonalcoholic steatohepatitis: association with obesity and insuline resistance, and influence weight loss. Diabetes Metab 2000; 26: 98-106. [ Links ]
33. García-Monzón C, Martínez-Pérez E, Iacono OL, Fernández-Bermejo M, Majano PL, et al. Characterization of pathogenic and prognostic factors of nonalcoholic steatohepatitis associated with obesity. J Hepatol 2000; 33: 716-24. [ Links ]
34. Matsuzawa Y, Shimomura I, Nakamura T, Keno Y, Kotani K, Tokunaga K. Pathophysiology and pathogenesis of visceral fat obesity. Obes Res 1995; 3 (Supl. 2): 187S-194S. [ Links ]
35. Cortez-Pinto H, Camilo ME, Baptista A, De Oliveira AG, De Moura MC. Non-alcoholic fatty liver: another feature of the metabolic syndrome? Clin Nutr 1999; 18: 353-835. [ Links ]
36. Fiatarone JR, Coverdale SA, Batey RG, Farrell GC. Non-alcoholic steatohepatitis impaired antipyrine metabolism and hypertriglyceridaemia may be clues to its pathogenesis. J Gastroenterol Hepatol 1991; 6: 585-90. [ Links ]
37. Reid AE. Nonalcoholic steatohepatitis. Gastroenterology 2001; 121: 710-23. [ Links ]
38. Pessayre D, Mansouri A, Fromenty B. Non alcoholic steatohepatitis (NASH): potential cause and pathogenic mechanisms. In: Hepatology 2000 (Falk Symposium Nº 117) Dordrecht: Kluwer Academic Publishers, 2001. p. 57-76. [ Links ]
39. Marchesini G, Brizi M, Bianchi G, Tomasselli S, Zoll M, Melchlonda N. Metformin in non-alcoholic steatohepatitis. Lancet 2001; 358: 893-4. [ Links ]
40. Sanyal, AJ, Campbell-Sargent C, Mirshashi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001; 120: 1183-92. [ Links ]
41. Ducluzeau PH, Fletcher LM, Vidal H, La Ville M, Tavaré JM. Molecular mechanisms of insulin-stimulated glucose uptake in adipocytes. Diabetes Metab 2002; 28: 85-92. [ Links ]
42. Kaplan LM. Leptin, obesity, and liver disease. Gastroenterology 1998; 115: 997-1001. [ Links ]
43. Chen J, Schenker S, Frosto TA, Henderson GI. Inhibition of cytochrome c oxidase activity by 4-hydroxynonenal (HNE). Role of HNE adduct formation with the enzyme catalytic site. Biochem Biophys Acta 1998; 1380: 336-44. [ Links ]
44. Pessayre D, Berson A, Fromenty B, Mansouri A. Mitochondria in steatohepatitis. Semin Liver Dis 2001; 21: 57-68. [ Links ]
45. Caldwell SH, Swerdlow RH, Khan EM, Iezzoni JC, Hespenheide EE, Parks JK. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J Hepatol 1999; 31: 430-4. [ Links ]
46. Pérez-Carrera M, Del Hoyo P, Martín M, Rubio J, Martín A, Garfia C, et al. Activity of the mitochondrial respiratory chain enzymes is decreased in the liver of patients with non-alcoholic steatohepatitis. Hepatology 1999; 30: 379A. [ Links ]
47. Cortez-Pinto H, Chatham J, Chaco VP, Arnold C, Rashid A, Diehl AM. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis. A pilot study. JAMA 1999; 282: 1659-64. [ Links ]
48. Chitturi S, Farrell GC. Etiopathogenesis of nonalcoholic steatohepatitis. Semin Liver Dis 2001; 21:27-41. [ Links ]
49. Pessayre C, Haouzi D, Fau D, Robin MA, Mansouri A, Berson A. Withdrawal of life support, altruistic suicide, fratricidal killing and euthanasia by lymphocytes: different forms of drug-induced hepatic apoptosis. J Hepatol 1999; 31: 760-70. [ Links ]
50. Takahashi T, Tanaka M, Inazawa J, Abe T, Suda T, Nagata S. Human Fas ligand: gene structure, chromosomal location and species specificity. Int Immunol 1994; 6: 1567-74. [ Links ]
51. Fernández-Real JM, Ricart Engel W, Arroyo E, Balanca R, Casamitjana R, Cabrero D, et al. Serum ferritin as a component of the insulin resistance syndrome. Diabetes Care 1998; 21: 62-8. [ Links ]
52. Cavallo Perin P, Pacini G, Cerutti F, Bessone A, Condo C, Sacchetti L, et al. Insulin resistance and hyperinsulinemia in homozygous beta-thalassemia. Metabolism 1955; 44: 281-6. [ Links ]
53. Tuomainen TP, Nyyssonen K, Salonen R, Tervahauta A, Korpela H, Lakka T, et al. Body iron stores are associated with serum insulin and blood glucose concentrations. Diabetes Care 1997; 20: 426-8. [ Links ]
54. Merkel PA, Simonson DC, Amiel SA, Plewe G, Sherwin RS, Pearson HA, et al. Insulin resistance and hyperinsulinemia in patients with thalassemia maior treated by hypertransfusion. N Engl J Med 1988; 318: 809-14. [ Links ]
55. Desai TK. Phlebotomy reduces transaminase levels in patients with non-alcoholic steatohepatitis (Abstract). Gastroenterology 2000; 118: A975. [ Links ]
56. Nitecki J, Jackson FW, Allen ML. Effect of phlebotomy on non-alcoholic steatohepatitis (NASH) (Abstract). Gastroenterology 2000; 118: A1474. [ Links ]
57. Mendler MH, Turlin B, Moirand R, Jouanolle AM, Sapey T, Guyader D, et al. Insulin resistance-associated hepatic iron overload. Gastroenterology 1999; 117: 1155-63. [ Links ]
58. Bonkovsky HL, Jawaid Q, Tortorelli K, Eclair P, Cobb J, Lambrecht RW. Non-alcoholic steatohepatitis and iron: increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J Hepatol 1999; 31: 421-9. [ Links ]
59. Siegmund B, Lehr HA, Fantuzzi G. Leptin: a pivotal mediator of intestinal inflammation in mice. Gastroenterology 2002; 122: 2011-25. [ Links ]
60. Anania FA. Leptin, liver, and obese mice-fibrosis in the fat lane. Hepatology 2002; 36: 246-8. [ Links ]
61. Ikejima K, Honda H, Yoshikawa M, Hirose M, Kitamura T, Takei Y et al. Leptin augments inflammatory and profibrogenic responses in the murine liver-induced by hepatotoxic chemicals. Hepatology 2001; 34: 288-97. [ Links ]
62. Ikejima K, Takei Y, Honda H, Hirose M, Yoshikawa M, Zhang YJ, et al. Leptin receptor-mediated signalling regulates hepatic profibrogenic and remodelling of extracellular matrix in the rat. Gastroenterology 2002; 122: 1399-410. [ Links ]
63. Diehl AM. Nonalcoholic steatosis and steatohepatitis IV. Nonalcoholic fatty liver disease abnormalities in macrophage function and cytokines. Am J Physiol Gastrointest Liver Physiol 2002; 282: G1-G5. [ Links ]
64. Shimomura I, Hammer RE, Ikemoto S, Brown MS, Goldstein JL. Leptin reverses insulin resistance and diabetes mellitus in mice with congenital lipodystrophy. Nature 1999; 401: 73-6. [ Links ]
65. Solga SF, Diehl AM. Non-alcoholic fatty liver disease: lumen-liver interactions and possible role of probiotics. J Hepatol 2003; 38: 681-7. [ Links ]
66. Malloy MJ, Kane JP, Bacon B. A genetic mutation in the proliferator activated receptor alpha (PPAR-A) gene in patients with non-alcoholic steatohepatitis. Hepatology 2002; 34: 441 (abstract A1076). [ Links ]
67. Baldridge AD, Pérez-Atayde AR, Graeme-Cook F, Higgins L, Lavine JE. Idiopathic steatohepatitis in childhood: a multicenter retrospective study. J Pediatr 1995; 127: 700-4. [ Links ]
68. Rashid M, Roberts EA. Nonalcoholic steatohepatitis in children. J Pediatr Gastroenterol Nutr 2000; 30: 48-52. [ Links ]
69. Sheth SG, Gordon FD, Chopra S. Non-alcoholic steatohepatitis. Ann Intern Med 1997; 126: 137-45. [ Links ]
70. Falck-Ytter Y, Younossi ZM, Marchesini G, McCullough J. Clinical features and natural history of nonalcoholic steatosis syndromes. Semin Liver Dis 2001; 21: 17-26. [ Links ]
71. Sanyal AJ. Non-alcoholic steatohepatitis. Clin Perspect Gastroenterol 2000: 129-39. [ Links ]
72. Kumar KS, Malet PF. Nonalcoholic steatohepatitis. Mayo Clin Proc 2000; 75: 733-9. [ Links ]
73. Leone N, Bugianesi E, Brunello F, Carucci P, Vanni E, Marchesini G, et al. Does nonalcoholic steatohepatitis progress to cryptogenic cirrhosis and hepatocellular carcinoma? A case-control study. (Abstract). Hepatology 2001; 34: (Part 2). 449 A. [ Links ]
74. Nair S, Eason JD, Loss GE, Mason AL. Relative risk of hepatocellular carcinoma in cirrhosis due to fatty liver disease: a study based on incidental hepatocellular carcinoma in liver transplant recipient. (Abstract). Hepatology 2001; 34 (Part 2): 462 A. [ Links ]
75. Bugianesi E, Leone N, Vanni E, Marchesini G, Brunello F, Caruchi P, et al. Expanding the natural history of nonalcoholic steatohepatitis: From cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology 2002; 123: 134-40. [ Links ]
76. Yang S, Lin HZ, Hwang J, Chacko VP, Diehl AM. Hepatic hyperplasia in non cirrhotic fatty livers: is obesity-related hepatic steatosis a premalignant condition? Cancer Res 2001; 61: 5016-23. [ Links ]
77. El-Serag HB, Richardson OA, Everhart JE. The role of diabetes in hepatocellular carcinoma. A case-control study among United States Veterans. Am J Gastoenterol 2001; 96: 2462-7. [ Links ]
78. George DK, Goldwurm S, MacDonald GA, Cowley LL, Walker NI, Ward PJ, et al. Increased hepatic iron concentration in non-alcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology 1998; 114: 311-8. [ Links ]
79. Moirand R, Mortaji AM, Loréal O, Paillard F, Brissot P, Deugnier Y. A new syndrome of liver iron overload with normal transferrin saturation. Lancet 1997; 349: 95-7. [ Links ]
80. Younossi ZM, Gramlich T, Bacon BR, Matteoni CA, Boparai N, O'Neill R, et al. Hepatic iron and nonalcoholic fatty liver. Hepatoloy 1999; 30: 847-50. [ Links ]
81. Fargion S, Mattioli M, Fracanzani AL, Sampietro M, Tavazzi D, Fociani P, et al. Hyperferritinemia, iron overload, and multiple metabolic alterations identify patients at risk for nonalcoholic steatohepatitis. Am J Gastroenterol 2001; 96: 2448-55. [ Links ]
82. Chitturi S, Weltman M, Farrell GC, McDonald D, Kench J, Liddle C et al. HFE mutations, hepatic iron, and fibrosis, ethnic-specific association of NASH with C262Y but not with fibrotic severity. Hepatology 2002; 36: 142-9. [ Links ]
83. Brunt EM. Nonalcoholic steatohepatitis: Definition and pathology. Semin Liver Dis 2001; 21: 3-16. [ Links ]
84. Joseph AE, Saverymuttu SH, al-Sam S, Cook MG, Maxwell JD. Comparison of liver histology with ultrasonography in assessing diffuse parenchymal liver disease. Clin Radiol 1991; 43: 26-31. [ Links ]
85. Matthews EM, Hosker JH, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and b-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28: 412-9. [ Links ]
86. Chitturi S, Abeygunasekera S, Farrell A, Holmes-Walker J, Hui JM, Fung C, et al. NASH and insuline resistance: insulin hypersecretion and specific association with the insulin resistance syndrome. Hepatology 2002; 35: 373-9. [ Links ]
87. Marchesini G, Forlani G. NASH: From liver diseases to metabolic disorders and back to clinical hepatology. Hepatology 2002; 35: 497-9. [ Links ]
88. Cobelli C, Pacini G. Insulin secretion and hepatic extraction in humans by minimal modelling of C-peptide and insulin kinetics. Diabetes 1988; 37: 223-31. [ Links ]
89. McCullough AJ. Update on nonalcoholic fatty liver disease. J Clin Gastroenterol 2002; 34: 255-62. [ Links ]
90. Chamberlain J, DeMouy J. Diet and exercise dramatically delay type 2 diabetes; diabetes medication, metformin, also effective. U.S. Department of Health and Human Services Press Release 200; 8-8-01. http:\\www.hhs.gov\news\press\2001press\200110808a.html. [ Links ]
91. Angulo P, Lindr KD. Treatment of nonalcoholic fatty liver: present and emerging therapies. Semin Liver Dis 2001; 21: 81-8. [ Links ]
92. Erikkson S, Eriksson KF, Bondesson L. Nonalcoholic steatohepatitis in obesity: a reversible condition. Acta Med Scand 1986; 220: 83-8. [ Links ]
93. Agrawal S, Bonkovsky HL. Management of nonalcoholic steatohepatitis. An analytic review. J Clin Gastroenterol 2002; 35: 253-61. [ Links ]
94. Basaranoglu M, Acbay O, Sonsuz A. A controlled trial of gemfibrozil in the treatment of patients with nonalcoholic steatohepatitis. J Hepatol 1999; 31: 384. [ Links ]
95. Laurin J, Lindor KD, Crippin JS, Gossard A, Gores GJ, Ludwig J, et al. Ursodeoxycholic acid or clofibrate in the treatment of nonalcoholic-induced steatohepatitis: a pilot study. Hepatology 1996; 23: 1464-7. [ Links ]
96. Salbara T, Onishi S, Ogawa Y, Yoshida S, Enzan H. Bezafibrate for tamoxifen-induced non-alcoholic steatohepatitis. Lancet 1999; 353: 1802. [ Links ]
97. Merat S, Malekzadeh R, Sohrabi MR, Sotoudeh M, Rakhshani N, Sohrabpour AA, et al. Probucol in the treatment of non-alcoholic steatohepatitis: a double-blind randomized controlled study. J Hepatol 2003; 38: 414-8. [ Links ]
98. Harlander JC, Kwo PY, Cummings OW. Atorvastin for the treatment of NASH. Gastroenteroloy 2001; 120A: 544-2767. [ Links ]
99. Lin HZ, Yang SQ, Kujhada F, Ronnet G, Diehl AM. Metformin reverses nonalcoholic fatty liver disease in obese leptin-deficient mice. Nat Med 2000; 6: 998-1003. [ Links ]
100. Kakuma T, Lee Y, Higa M, Wang ZW, Pan W, Shimomura I, et al. Leptin, troglitazone, and the expression of sterol regulatory element binding proteins in liver and pancreatic islets. Proc Natl Acad Sci USA 2000; 97: 8536-41. [ Links ]
101. Caldwell SH, Hespenheide EE, Redick JA, Iezzoni JC, Battle EH, Sheppard BL. A pilot study of a thiazolidinedione, troglitazone, in nonalcoholic steatohepatitis. Am J Gastroenterol 2001; 96: 519-25. [ Links ]
102. Newschwander-Tetri BA, Brunt EM, Wehmeier KR, Oliver D, Bacon BR. Improved nonalcoholic steatohepatitis after 48 weeks of treatment with PPAR gamma ligand rosiglitazone. Hepatology 2003; 38: 1008-17. [ Links ]
103. Bajaj M, Suraamornkul S, Piper P, Hardies LJ, Glass L, Cersosimo E, et al. Decreased plasma adiponectin concentration are closely correlated to hepatic fat content and hepatic insuline resistance in pioglitazone-treated type 2 diabetic patients. J Clin Endocrinol Metab 2004; 89: 200-6. [ Links ]
104. Hookman P, Barkin JS. Current biochemical studies of non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) suggest a new therapeutic approach. Am J Gastroenterol 2003; 98: 495-9. [ Links ]
105. Guma G, Viola L, Thome M. Ursodeoxycholic acid in the treatment of nonalcoholic steatohepatitis: results of a prospective clinical controlled trial (Abstract). Hepatology 1997; 26: 387A. [ Links ]
106. Ceriani R, Bunati S, Morini L. Effect of ursodeoxycholic acid plus diet in patients with non-alcoholic steatohepatitis (Abstract). Hepatology 1998; 28: 368A. [ Links ]
107. Lindor KD, Kowdley V, Heathcote EJ, Harrison ME, Jorgensen R, Angulo P, et al. Ursodeoxycholic acid for treatment of nonalcoholic steatohepatitis: Results of a randomized trial. Hepatology 2004; 39: 770-80. [ Links ]
108. Nanji AA, Yang EK, Fogt F, Sadrzadeh SM, Dannenberg AJ. Medium chain triglycerides and vitamin E reduce the severity of established experimental alcoholic liver disease. J Pharmacol Exp Ther 1996; 277: 1694-700. [ Links ]
109. Lavine JE. Vitamin E treatment of non-alcoholic steatohepatitis in children: a pilot study. J Pediatr 2000; 136: 734-8. [ Links ]
110. Haseagawa T, Yoneda M, Nakamura K, Yokihama S, Tamonk K, Sato Y. Diagnostic significance of measuring TGF-a1 levels and effect of a-tocoferol in patients with NASH. Gastroenterology 1997; 112: 1278. [ Links ]
111. Barak AJ, Beckenhauer HC, Badakhsh S, Tuma DJ. The effect of betaina in reversing alcoholic steatosis. Alcohol Clin Exp Res 1997; 21: 1100-2. [ Links ]
112. Abdelmalek MF, Angulo P, Jorgensen RA, Sylvestre PB, Lindor KD. Betaine, a promising new agent for patients with nonalcoholic steatohepatitis: results of a pilot study. Am J Gastroenteol 2001; 96: 2711- 7. [ Links ]
113. Velusssi M, Cernigoi AM, De Monte A, Dapas F, Caffau C, Zilli M. Long term (122 months) treatment with an anti-oxidant drug is effective on hyperinsulinemia, exogenous insulin need, and malondialdehyde in cirrhotic diabetic patients. J Hepatol 1997; 26: 871-9. [ Links ]
114. Gulbahar O, Karusi ZA, Ersoz G. Treatment of non-alcoholic steatohepatitis with N-acetylcysteine. Gastroenterology 2000; 118: A 1444. [ Links ]
115. Pappo I, Becovier H, Berry EM, Freund HR. Polymixin B reduces cecal flora. TNF production and hepatic steatosis during total parenteral nutrition in rat. J Clin Res 1991; 51: 106-12. [ Links ]
116. Freund HR, Muggia-Sullan M, LaFrance R, Enrione EB, Popp HB, Bjornson HS. A possible beneficial effect of metronidazole in reducing TPN-associated liver function derangements. J Surg Res 1985; 38: 356-63. [ Links ]
117. Buchman AL, Dubin MD, Moukarzel AA, Jenden DJ, Roch M, Rice KM, et al. Choline deficiency: a cause of hepatic steatosis during parenteral nutrition that can be reversed with intravenous choline supplementation. Hepatology 1995; 22: 1399-403. [ Links ]
118. Fernández-Real JM, López-Bermejo, Ricart W. Cross-talk between iron metabolism and diabetes. Diabetes 2002; 51: 2348-54. [ Links ]
119. Guillyomarc'h A, Mendler R, Moirand R, Lainé F, Quentin V, David V, et al. Venesection therapy of insulin resistence-associated hepatic iron overload. J Hepatol 2001; 35: 344-9. [ Links ]
120. Fachini FS, Hua NW, Stoohs RA. Effect of iron depletion in carbohydrate-intolerant patients with clinical evidence of non-alcoholic liver disease. Gastroenterology 2002; 122: 931-9. [ Links ]
121. Kim W, Poterucha J, Porayko M, Dickson E, Steers J, Wiesner R. Recurrence of nonalcoholic steatohepatitis following liver transplantation. Transplantation 1996; 62: 1802-5. [ Links ]
122. Carlson K, Washington M, Treem W, Clavien P, Hunt C. Recurrence of nonalcoholic steatohepatitis in a liver transplant recipient. Liver Transpl Sur 1997; 3: 174-6. [ Links ]
123. Molloy R, Komorowski R, Varma R. Recurrent nonalcoholic steatohepatitis and cirrhosis after liver transplantation. Liver Transpl Sur 1997; 3: 177-8. [ Links ]
124. Selzner M, Clavien P-A. Fatty liver in liver transplantation and surgery. Semin Liver Dis 2001; 21: 105-13. [ Links ]

