










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.97 no.8 Madrid ago. 2005
Cartas al Director
Penetrancia familiar del gen HFE: los cuatro hermanos de una familia afectados por hemocromatosis hereditaria
Palabras clave: Gen HFE. Hemocromatosis. Penetrancia.
Key words: HFE gene. Hemochromatosis. Penetrancy.
Sr. Director:
La hemocromatosis hereditaria (HH) es un trastorno metabólico de base genética que conduce a una sobrecarga de hierro en el organismo. La mayoría de los pacientes son homozigotos para el alelo C282Y del gen HFE, otros casos son "heterozigotos compuestos" (C282Y/H63D) y un pequeño porcentaje de enfermos no presenta estas alteraciones, por lo que se han involucrado otras posibles mutaciones (S65C y otras). En ciertas áreas, la relevancia de la mutación C282Y en la HH no es tan evidente como en Europa o Norteamérica, existiendo diferencias étnicas en la distribución de estas mutaciones. Además el exceso de hierro varía con la edad, el sexo, la historia previa de donaciones de sangre, la ingesta de alcohol y otros factores. Para complicar más el panorama se ha descrito que la homozigosis C282Y no siempre implica el desarrollo de la enfermedad fenotípica habiendo numerosos estudios poblacionales que muestran resultados muy discordantes respecto a la penetrancia de la mutación. Probablemente la HH se comporta como una enfermedad con herencia autosómica recesiva y penetrancia incompleta. Por otra parte otras muchas causas distintas de la HH pueden producir sobrecarga férrica, no existiendo un consenso absoluto sobre la nomenclatura a utilizar.
Se presenta el caso de una familia en la que los cuatro hermanos presentaban HH siendo relevante por su diagnóstico casual, su expresión genotípica y fenotípica prácticamente idéntica con datos de sobrecarga férrica intensa, la baja probabilidad de aparición en la población general y el importante beneficio obtenido (diagnóstico y tratamiento tempranos) gracias a una adecuada y exhaustiva valoración de los casos. Se trata de cuatro varones jóvenes que fueron estudiados en nuestra consulta después de diagnosticar a uno de ellos de HH tras haberse investigado una elevación del hierro de una analítica rutinaria. En todos se realizó una anamnesis completa, una exploración física detallada y varias pruebas complementarias: determinación de las mutaciones C282Y y H63D del gen HFE, hemograma, bioquímica completa con perfil hepático, estudio de coagulación, determinación de hierro, de ferritina y del índice de saturación de transferrina (IST), inmunoglobulinas, serologías virales, porfirinuria, anticuerpos no organoespecíficos, ecografía abdominal, biopsia hepática con cuantificación de hierro, índice de hierro hepático (IHH) y radiografías de manos y rodillas (Tabla I). Finalmente los cuatro fueron diagnosticados de HH y se comenzó con flebotomías periódicas, presentando buena respuesta analítica y permaneciendo asintomáticos hasta la fecha.
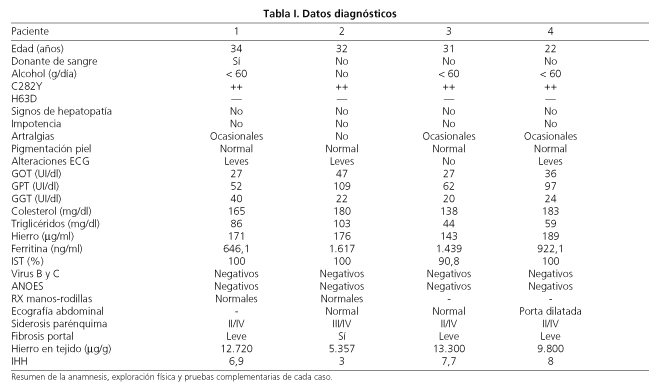
Tras la realización de las pruebas diagnósticas se observó que los cuatro hermanos eran homozigotos C282Y por lo que también se estudió a los padres, objetivándose que eran heterozigotos para el C282Y y no padecían la enfermedad. La probabilidad calculada de que en los 4 hijos existiera este genotipo era bastante baja (0,39%). Respecto al diagnóstico fenotípico se observa cómo en todos los casos iban apareciendo diversas alteraciones clínicamente leves pero que ya indicaban el desarrollo de la enfermedad: artralgias ocasionales, transaminasas discretamente elevadas, mínimas alteraciones en el ECG, elevación de los parámetros relacionados con el hierro, siderosis parenquimatosa y fibrosis portal en evolución. No había datos de hepatopatía avanzada, cardiopatía severa, impotencia o hiperpigmentación. Sin duda, el diagnóstico temprano fue fundamental para evitar posteriores complicaciones. Esto indica la importancia y dificultad del diagnóstico precoz y la obligación de realizar un adecuado estudio en familiares de primer grado de los casos conocidos.
Otro punto importante es la expresión fenotípica de la homozigosis C282Y, que en esta familia es muy evidente como lo demuestran los síntomas y los parámetros relacionados con el hierro, como la cantidad de hierro en tejido hepático (> 5.000 µg/g en todos los casos) y el específico IHH. Además no había factores de riesgo "ferrogénico" (hiperlipidemias, ingesta excesiva de alcohol, hepatitis, porfirias o esteatosis) que influyeran en el almacenamiento de hierro, por lo que hubo pocas dudas diagnósticas. Sin embargo, en la práctica habitual no todos los casos son tan evidentes ya que hay veces en las que individuos con sobrecarga férrica moderada, alguna de las mutaciones y/o algún factor "ambiental" añadido pueden ser diagnosticados erróneamente de HH, al igual que hay pacientes con enfermedad fenotípica no diagnosticada debido a su joven edad, al efecto protector de la menstruación o de las donaciones o a la falta de sospecha.
Beutler y cols. (1) recientemente realizaron un screening de estas mutaciones en 41.038 individuos y observaron que sólo un 1% de los homozigotos C282Y presentaba la enfermedad. Son unos resultados muy llamativos, similares a los reportados por McCune y cols. (2) al estudiar una población galesa. Sin embargo, hay estudios que presentan una expresión fenotípica muy superior y otros autores (3,4) discuten estos datos e insisten en que hay que dejar muy claros los criterios fenotípicos (clínico-analíticos) de la HH. Phatak y cols. (5) analizaron 4.865 casos detectando 12 C282Y/C282Y: el 83% tenían un IST > 55%, el 42% una ferritina > 300 ng/ml y no hubo ningún caso de cirrosis o cardiopatía. Parece necesario seguir investigando este tema.
En nuestro caso, con una penetrancia del 1%, la probabilidad de que 4 personas con el genotipo C282Y/C282Y presentaran la enfermedad clínica sería de 1 entre 100 millones, lo que obliga a reflexionar sobre si es un caso excepcional, si la penetrancia real es más elevada o si se podría plantear la posibilidad de que exista algún otro factor genético-familiar añadido que favorezca el desarrollo de la enfermedad cuando se presenta este genotipo. Barton y cols. (6) estudiaron a los familiares de 61 enfermos de HH y detectaron 25 casos homozigotos C282Y, de los cuales 23 (92%) presentaban la enfermedad fenotípica, hasta entonces no diagnosticada. Por otra parte, Adams y cols. (7) afirman que muchos homozigotos C282Y no tendrán sobrecarga clínica de hierro, aunque el riesgo es mayor en hombres y en los hermanos varones homozigotos.
La influencia real del gen HFE, el papel de otros factores ambientales en el desarrollo de la enfermedad fenotípica, la existencia de posibles determinantes genéticos no conocidos, la adecuada caracterización de los síndromes de sobrecarga férrica y la relación con las mutaciones del gen HFE son temas que deben investigarse aún más. Es esencial realizar siempre un estudio detallado de los casos sospechosos.
M. Vázquez Romero, D. Boixeda de Miquel, C. Martín de Argila de Prados,
I. Vallcorba Gómez del Valle1, P. Cabello Albendea1, A. López San Román
y C. San Román Cos-Gayón1
Servicios de Gastroenterología-Hepatología y 1Genética.
Hospital Ramón y Cajal. Madrid
Bibliografía
1. Beutler E, Felliti VJ, Koziol JA, Ho NJ, Gelbart T. Penetrance of 845G-A (C282Y) HFE hereditary haemochromatosis mutation in the USA. Lancet 2002; 359: 211-8.
2. McCune CA, Al-Jader LN, May A, Hayes SL, Jackson JA, Worwood M. Hereditary haemochromatosis: only 1% of adult HFE-C282Y homozygotes in South Wales have a clinical diagnosis of iron overload. Hum Genet 2002; 111: 538-43.
3. Poullis A, Moodie SJ, Maxwell JD. Clinical haemochromatosis in HFE mutation carriers. Lancet 2002; 360: 411-2.
4. Basset ML, Wilson SR, Cavanaugh JA. Penetrance of HFE-related hemochromatosis in perspective. Hepatology 2002; 36: 500-3.
5. Phatak PD, Ryan DH, Cappuccio J, Oakes D, Braggins C, Provenzano K, et al. Prevalence and penetrance of HFE mutations in 4865 unselected primary care patients. Blood Cells Mol Dis 2002; 29: 41-7.
6. Barton JC, Rothenberg BE, Bertoli LF, Acton RT. Diagnosis of hemochromatosis in family members of probands: a comparison of phenotyping and HFE genotyping. Genet Med 1999; 1: 89-93.
7. Adams PC, Walker AP, Acton RT. A primer for predicting of disease in HFE-linked hemochromatosis. Genet Test 2002; 5: 311-6.

