










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.99 no.7 Madrid jul. 2007
PUNTO DE VISTA
Mecanismos de lesión hepatocelular
Mechanisms of liver injury
J. Muntané, R. González, I. Ranchal, J. A. Collado, L. M. López-Sánchez, C. Herencia,
A. Rodríguez-Ariza, J. R. Muñoz-Castañeda y M. de la Mata
Liver Research Unit. Reina Sofía University Hospital. Ciberehd. Córdoba
Dirección para correspondencia
Muerte celular
La muerte celular es un proceso que acompaña a numerosas situaciones fisiológicas y patológicas en los organismos. El primer patrón de muerte celular identificado fue la necrosis celular, descrita por Virchow en 1871. Posteriormente se observó que la muerte celular formaba parte integrada de los mecanismos normales de diferenciación celular y tisular de los organismos superiores. En este sentido, los primeros estudios de embriología constataron que los procesos de muerte celular eran necesarios para modelar la forma final de los organismos. La morfogénesis conlleva sistemáticamente la eliminación y generación de nuevas estructuras celulares y tisulares. Un fenómeno similar se encuentra durante la metamorfosis en invertebrados y vertebrados inferiores en donde la masiva involución tisular y la eliminación celular son procesos fisiológicos que se desarrollan de forma coordinada. Este proceso de muerte celular denominado apoptosis fue caracterizado por Kerr en 1965. La apoptosis y necrosis celular se distinguen por una serie de parámetros morfológicos y bioquímicos. La apoptosis consiste en la eliminación controlada de la célula afectada sin alterar de forma relevante el metabolismo celular. Este proceso se caracteriza por la activación secuencial de una serie de proteasas denominadas caspasas que actúan sobre las uniones cisteína-aspartato del substrato. La activación de las caspasas conlleva la fragmentación del DNA y la alteración de la arquitectura celular, que se acompaña de una serie de cambios morfológicos como la condensación del DNA nuclear, disminución del volumen celular y generación de cuerpos apoptóticos sin liberación del contenido intracelular. La necrosis resulta de la perturbación extrema del equilibrio celular que afecta el metabolismo celular con disminución drástica del contenido energético celular en forma de adenosina trifosfato (ATP), alteración del contenido iónico, incremento del volumen mitocondrial y celular, y con activación de proteasas intracelulares. Este proceso culmina con la ruptura de la membrana celular y liberación del contenido celular que promueve una reacción inflamatoria secundaria. En el hígado la presencia de apoptosis celular tiende a presentar una distribución focal, mientras que la necrosis hepatocelular presenta una distribución zonal. A pesar de la distinción clara existente entre apoptosis y necrosis, en el hígado suelen coexistir ambos tipos de muerte celular por el hecho que un mismo estímulo es capaz de inducir apoptosis o necrosis según el tipo celular afectado, el grado de exposición, el estado metabólico celular y la integridad de la maquinaria que participa en la muerte celular. En este sentido, diversos autores han sugerido que apoptosis y necrosis no son dos procesos separados sino que son extremos opuestos de un mismo mecanismo celular denominado necrapoptosis (1). El contenido celular de ATP generado por la mitocondria es un factor clave que regula la inducción de apoptosis o necrosis en el proceso de muerte celular. En este sentido, una lesión que afecta a pocas mitocondrias puede ser resuelta por autofagia de los orgánulos alterados. Si en el proceso participan más mitocondrias que liberan una cantidad suficiente de factores pro-apoptóticos pero manteniéndose los niveles de ATP suficientes la célula entra en apoptosis. Si la célula sufre una lesión severa, la drástica reducción del contenido en ATP no permite el mantenimiento de numerosos procesos dependientes de energía como el control de la ósmosis celular y la permeabilidad de la membrana celular, lo que induce la muerte celular por necrosis. Es importante resaltar que no existe un parámetro, con excepción de la morfología celular in vivo, que sea específico de la apoptosis celular. En este sentido, son necesarios varios parámetros cuantitativos y cualitativos para determinar cuál es el tipo de muerte celular implicada en una situación fisiopatológica concreta.
Mecanismo de muerte celular
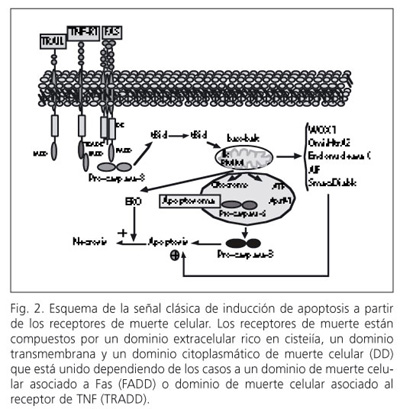
Las caspasas son una familia de proteasas presentes en la célula en forma inactiva o zimógenos que se activan de forma secuencial por la acción de otras caspasas activas. La activación de caspasas puede ocurrir mediante 2 procesos denominados intrínseco y extrínseco que dependen del tipo y del estado fisiopatológico celular. En el primer mecanismo, señales generadas intracelularmente (p. ej., Bax, Bak, Bid, Puma, Noxa, VpR, p53, JNK, Ca2+, especies reactivas de oxígeno o ERO, ceramida, gangliósido GD3, etc.) actúan en la mitocondria induciendo la liberación de señales de muerte celular. En la vía extrínseca, la inducción de muerte celular se inicia mediante la unión de una serie de ligandos (TNF-a, Fas, TRAIL, etc.) a sus correspondientes receptores que inducen la agrupación de diversas proteínas adaptadoras y zimógenos de caspasas denominadas iniciadoras (procaspasa-8 ó 10) en el dominio intracelular del receptor para formar el denominado complejo de señal inductora de muerte (DISC) que transmite la señal de muerte celular directamente, o en la mayor parte de las situaciones mediante factores pro-apoptóticos liberados por la mitocondria que inducen la activación de caspasas ejecutoras (caspasa -3, -6 y -7) (Fig. 1). En ambos casos, la permeabilización de la membrana mitocondrial (PMM) es un acontecimiento crucial que tiene lugar tanto en la muerte celular por apoptosis como necrosis. Se han descrito dos mecanismos principales de inducción de PMM dependiendo del tipo celular, estímulo, intensidad y duración. En el primero, factores como Bid o Bax, tras su translocación y oligomerización en la membrana mitocondrial externa, forman poros para permitir la liberación de citocromo c o Smac/DIABLO del espacio intermembrana al citoplasma celular manteniendo la integridad de la membrana interna mitocondrial. La formación de estos canales se facilita por la incorporación de moléculas lipídicas (ceramida, esfingosina y gangliósido GD3) que inducen la generación de poros de mayor tamaño en respuesta a estímulos pro-apoptóticos. Por otra parte, la inducción del estado de transición de la permeabilidad mitocondrial (MPT) constituye un modelo alternativo a la formación de canales específicos en la membrana externa mitocondrial (2). En el proceso de la MPT participa un complejo multiproteico denominado canal aniónico dependiente de voltaje (VDAC) localizado en los puntos de contacto entre la membrana interna y externa mitocondrial, citocromo c localizado en la cara externa de la membrana interna mitocondrial, así como el estado del potencial de la membrana interna mitocondrial (Fig. 1). VDAC está constituido por una porina que es una proteína formadora de poros, la proteína translocadora de ATP/ADP (ANT), la hexoquinasa que transforma la glucosa en glucosa-6-fosfato enzima iniciadora de la glucólisis, creatinina quinasa, diversos miembros anti-apoptóticos de la familia de Bcl-2 (Bcl-2 y Bcl-XL) y la ciclofilina D (Fig. 1). Durante la inducción de MPT tiene lugar la alteración del complejo VDAC y el colapso del potencial mitocondrial por despolarización de la cadena electrónica mitocondrial, el desacoplamiento de la fosforilación oxidativa, generación de ERO y la disipación de gradientes iónicos que conlleva la aparición de un poro de permeabilidad transitoria (PTP) que permitirá la salida de moléculas de hasta un peso molecular de 1,5 kDa. Sin embargo, debido al superior tamaño de los factores pro-apoptóticos intermembrana (citocromo c, AIF, pro-caspasa-9, etc.) se ha sugerido la presencia de grandes poros facilitados por las interacciones Bid-Bak o VDAC-Bax que conlleva la rotura de la membrana externa como consecuencia del incremento de volumen de la matriz mitocondrial y liberación de factores pro-apoptóticos del espacio intermembrana. La liberación de los factores pro-apoptóticos integrantes de la cadena electrónica mitocondrial, como el citocromo c, bien a través de PMN o MPT tiene dos consecuencias importantes que son la interrupción de la transferencia de electrones en el complejo III con la consecuente generación de ERO (3), y el inicio de la señal de apoptosis celular. La inducción de estrés oxidativo es esencial en la progresión de la disfunción mitocondrial. La oxidación de la cardiolipina, fosfolípido aniónico presente exclusivamente en algunos de los complejos de transporte electrónico mitocondrial, favorece la inducción y progresión de de la disfunción mitocondrial. Es probable que la generación de amplios poros de la membrana externa mitocondrial o la inducción de MPT formen parte de un mismo proceso común de alteración mitocondrial en el proceso de muerte celular. En este sentido, se ha observado que GD3 (4) y MPT (5) participan en la inducción de muerte celular por TNF-a en hepatocitos precediendo a la liberación de citocromo c, activación de caspasa-3, fragmentación del DNA y los cambios morfológicos asociados a los procesos apoptóticos. Asimismo, el hecho de la capacidad de Bid de interactuar con componentes generadores de poros en la membrana externa (Bak, Bax) o con componentes de MPT (Bcl2) sugiere la participación integrada de ambos tipos de procesos en la inducción de muerte celular. La señal de apoptosis clásica inducida por PMN o más específicamente por la MPT conlleva la liberación desde el espacio intermembrana mitocondrial del citocromo c, la pro-caspasa-9 y ATP que junto con el factor citosólico apaf-1 constituyen el apoptosoma (Fig. 2). La formación del apoptosoma induce la activación de la caspasa-9 que a su vez, mediante una proteolisis controlada de la procaspasa-3, da lugar a su activación que actúa sobre dianas específicas que determinan el fenotipo apoptótico. La inducción de necrosis celular es consecuencia de una amplia PMM que contribuye a la disfunción metabólica, pérdida brusca de ATP e inhabilitación para el ensamblaje del apoptosoma y progresión de la apoptosis celular. Los pasos posteriores a la activación de la caspasa-3 consisten en la activación de la ADNasa activada por caspasa (CAD) mediante la degradación e inactivación de su inhibidor constitutivo (ICAD), lo que da lugar a la fragmentación del ADN. Aunque la formación del apoptosoma es la vía clásica de fragmentación del DNA, la mitocondria libera otras proteínas localizadas en el espacio intermembrana que promueven la muerte celular, como el factor de inducción de la apoptosis (AIF) y la endonucleasa G, que inducen la rotura del ADN sin participación de las caspasas; así como de Smac/DIABLO y Omi/HtrA2 que promueven la muerte celular mediante la inactivación de los denominados inhibidores de apoptosis (IAPs) (Fig. 2).
Muerte celular en enfermedades hepáticas
Los modelos experimentales de lesión hepatocelular in vivo o in vitro son útiles para comprender los procesos de muerte celular e identificar nuevas dianas terapéuticas en las enfermedades hepáticas en humanos. Sin embargo, la relevancia de estos resultados obtenidos en animales de experimentación debe ser contrastada en las diversas situaciones clínicas de hepatopatía. En las siguientes secciones se describen los mecanismos de muerte celular en diversos procesos relevantes de disfunción hepática tales como la lesión por isquemia-reperfusión, esteatohepatitis alcohólica y no alcohólica, enfermedades colestáticas, hepatitis viral y carcinoma hepatocelular.
Lesión por isquemia-reperfusión
La lesión hepática por anoxia o hipoxia está causada por una absoluta o relativa deficiencia en oxígeno. La anoxia ocurre durante la trombosis de la arteria hepática durante el trasplante hepático ortotópico, el mantenimiento del órgano para el trasplante o durante la interrupción del flujo vascular portal durante la resección hepática (maniobra de Pringle). La hipoxia hepática ocurre durante reducciones del flujo sanguíneo como en la enfermedad veno-oclusiva, la necrosis centrolobulillar congestiva, lesión hepática alcohólica, casos de hipotensión y en shock hemorrágico. Asimismo, la lesión por reoxigenación o reperfusión es consecuencia de la entrada de sangre oxigenada en el tejido hepático después de un periodo de anoxia o hipoxia. El grado de lesión tisular tras la reperfusión depende de la existencia previa de un proceso anóxico o hipóxico, así como del tipo celular y la temperatura de isquemia.
Las primeras alteraciones durante la anoxia celular ocurren en la mitocondria. La falta de oxígeno como aceptor final de electrones en la cadena de transporte electrónico mitocondrial incrementa el estado reducido del orgánulo como consecuencia del aumento de la relación NADH:NAD+ y disminución de la generación de ATP a partir de ADP. En hepatocitos con niveles importantes de glucógeno para la glicolisis anaeróbica, el estado nutricional es un importante factor para la lesión por anoxia celular. Durante la anoxia celular, y de forma independiente a la disminución de la generación oxidativa de ATP, existen mecanismos intracelulares que conllevan a la inhibición de la ATP sintasa y al mantenimiento del gradiente iónico que preservan el potencial y la integridad mitocondrial. Esta respuesta adaptativa celular a la anoxia parece ser consecuencia de la liberación de calcio mitocondrial en hepatocitos en cultivo (6). Si la anoxia se revierte cuando el potencial mitocondrial no se ha alterado, el hepatocito recupera la plena funcionalidad mitocondrial y se previene la muerte celular. Si el periodo de anoxia es excesivo, se induce PMM y disfunción mitocondrial que conduce a la progresión del proceso de muerte celular. En este sentido la alteración de PMM tiene un papel más relevante que la depleción de la generación oxidativa de ATP en la lesión hepatocelular por anoxia. La susceptibilidad de las distintas células hepáticas a la lesión por anoxia no se relaciona con el grado de depleción intracelular de ATP sino con el grado de actividad proteasa intracelular. Los hepatocitos son las células con mayor actividad proteasa intracelular y más susceptibilidad en el proceso de anoxia celular. Aunque la lesión hepática por anoxia puede presentarse en diversos síndromes de tipo isquémico, la patología asociada a hipoxia es la que se encuentra de forma más frecuente en la práctica clínica diaria. Las condiciones celulares de baja tensión de oxígeno inducen la disminución de la actividad de algunos complejos mitocondriales y síntesis de ATP acompañado de desacoplamiento de la cadena electrónica mitocondrial y generación de ERO. En este sentido, los hepatocitos localizados en la zona II del acino hepático sometidos a hipoxia pierden más rápidamente la viabilidad que las células localizadas en la zona I y III (7).
La lesión por reperfusión es causada por la reintroducción de concentraciones fisiológicas de oxígeno en células previamente expuestas a condiciones no letales de hipoxia o anoxia. En estas condiciones, se generan grandes cantidades de ERO tanto en la mitocondria de hepatocitos como en las células de Kupffer, endoteliales o epiteliales de los conductos biliares. En el hepatocito, la re-entrada de oxígeno en presencia de una reducida actividad de los complejos electrónicos mitocondriales genera un incremento de la autoxidación del complejo I y III mitocondrial con la consecuente generación de ERO. Los hepatocitos son menos sensibles al estrés oxidativo tras reoxigenación comparado con las células endoteliales o epiteliales ductales debido al favorable equilibrio intracelular entre el estrés oxidativo y el contenido de antioxidantes. La lesión de las células endoteliales es crítica para la supervivencia del hígado expuesto a isquemia fría en el trasplante hepático pues la disrupción de los vasos reduce el flujo sanguíneo y en último término exacerba la necrosis hepatocelular y fallo del injerto. Asimismo, la activación de las células de Kupffer y estrelladas durante la reperfusión genera ERO, citoquinas pro-inflamatorias y otros factores quimiotácticos que contribuyen al daño post-isquémico, respuesta inflamatoria sistémica y al fallo multiorgánico que acompaña a la lesión isquémica del hígado.
Esteatohepatitis alcohólica y no alcohólica
La esteatohepatitis alcohólica (EHA) y no alcohólica (EHNA) presentan características histológicas hepáticas similares aunque la etiología primaria es diferente. En ambas situaciones se observa esteatosis hepática, inflamación y muerte hepatocelular con una marcada susceptibilidad al estrés oxidativo y la fibrosis que puede evolucionar a cirrosis y carcinoma hepatocelular. Se considera que la acumulación de lípidos en el citoplasma de hepatocitos es el primer estadio en la generación de EHA y EHNA. La regulación de los genes relacionados con la síntesis de ácidos grasos y colesterol está bajo el control de una familia de factores de transcripción denominados proteínas de unión a elementos reguladores de esteroides (SREBPs). Diversos trabajos demuestran que el alcohol incrementa la expresión de SREBPs y los genes implicados en el metabolismo lipídico en hepatocitos en cultivo y en el hígado de ratas alimentadas con una dieta rica en alcohol (8). Asimismo, la inducción de diabetes experimental asociada a insulina resistencia y obesidad se relaciona con un incremento de la expresión de SREBPs y de ácidos grasos en hígado (9). La acumulación de lípidos en los hepatocitos con EHA y EHNA promueve el estrés oxidativo, inflamación, muerte celular y fibrosis que se corresponde con un segundo estadio en la progresión de la hepatopatía. Los mediadores implicados en el desarrollo de EH y EHNA son en la mayoría de los casos coincidentes. En este sentido, TNF-a es una citoquina que se relaciona con la progresión de EHA y EHNA. El consumo de alcohol induce una marcada reducción de la capacidad de transporte de GSH a la mitocondria que incrementa la susceptibilidad del hepatocito a la inducción de estrés oxidativo y muerte celular por TNF en hepatocitos (10). Asimismo, los niveles de TNF-a circulantes de los pacientes con EHNA se relacionan con una mayor disfunción y estrés oxidativo mitocondrial en hepatocitos (11). Recientemente se ha observado en modelos nutricionales y genéticos de esteatosis hepática caracterizados por un incremento de triglicéridos, ácidos grasos o colesterol, que la acumulación de colesterol en mitocondria reduce el transporte de GSH a la mitocondria en hepatocitos por un mecanismo similar al descrito en el caso de EHA que determina una mayor susceptibilidad de estos hepatocitos a la muerte celular por TNF-a. La disfunción mitocondrial y el estrés oxidativo se asocian a un incremento de los parámetros de muerte celular en los hepatocitos. El estrés oxidativo es un factor determinante en la progresión de la fibrogénesis hepática en EHA y EHNA.
Enfermedad hepática por colestasis
La disminución de la secreción de sales biliares por los hepatocitos es común en las colestasis inducidas por drogas, cirrosis biliar primaria, colangitis esclerosante primaria y obstrucción biliar. Aunque la lesión inicial en los conductos biliares es de tipo inmunológico, tóxico o genético en muchas enfermedades hepatobiliares, la lesión hepatocelular se exacerba por un efecto directo citotóxico de las sales biliares hidrofóbicas en los hepatocitos. Esta citotoxicidad se caracteriza desde el punto de vista histológico por la presencia de marcadores relacionados con la muerte celular por apoptosis y necrosis en el hígado colestático. La muerte celular por sales biliares hidrofóbicas se asocia con alteración de la funcionalidad mitocondrial, siendo de tipo apoptótico o necrótico en función de la concentración del citotóxico. A pesar de la actividad detergente de las sales biliares, se reconoce que la concentración requerida para su actividad pro-apoptótica es inferior a la concentración crítica requerida para alterar la estructura micelar de las mitocondrias. La señal pro-apoptótica se inicia con la capacidad de las sales biliares hidrofóbicas de inducir agregación del receptores de tipo Fas or TRAIL con la consiguiente activación de caspasa-8, translocación de Bid/Bax a la mitocondria y generación de PMN y liberación de factores pro-apoptóticos en hepatocitos (12). Asimismo, las sales biliares inhiben la actividad de los complejos mitocondrial y la generación de ATP celular que inducen un incremento de la concentración intracelular de Ca2+ y activación de diversas proteasas proapoptóticas celulares en hepatocitos (12). La inducción de PMM por sales biliares hidrófobas se previene por ciclosporina A lo que sugiere que su citotoxicidad está mediada por mecanismos específicos de generación de amplios canales en la membrana interna mitocondrial. Las sales biliares incrementan el estrés oxidativo intracelular con amplificación de la señal de la PMM que puede inducir la progresión de la muerte celular de tipo necrótico en hepatocitos.
Hepatitis virales
La hepatitis vírica causada por virus B (HBV) o C (HCV) representa un grave problema de salud pública debido al gran número de pacientes infectados que en un elevado porcentaje van a progresar a cirrosis hepática y carcinoma hepatocelular (HCC). La persistente infección comporta un reiterado proceso de destrucción y regeneración tisular que supone un incremento del riesgo de desarrollar HCC. La lesión hepática por HBV y HCV está principalmente mediada por la respuesta inmune del huésped frente a los hepatocitos infectados que expresan en su membrana proteínas virales asociados a antígenos de histocompatibilidad de clase I. Los linfocitos T citotóxicos reconocen a la célula infectada e induce su muerte celular por apoptosis. Los cuerpos apoptóticos, o previamente denominados acidófilos o de Councilman, están presentes en el tejido hepático procedente de pacientes con hepatitis viral. Diversos estudios han demostrado que los linfocitos T citotóxicos expresan ligandos de Fas que participan en la inducción de apoptosis del hepatocitos infectado a través del receptor de Fas. Se ha observado la expresión de Fas en los hepatocitos y de Fas ligando en el infiltrado de células inflamatorias en el hígado de pacientes infectados por HCV (13) y HBV (14) que se correlaciona con la severidad de la lesión hepática. La expresión de Fas en los hepatocitos infectados puede ser inducida por proteínas virales específicas o por citoquinas inflamatorias como la interleucina-1 generada durante la respuesta inflamatoria. Otros mecanismos de muerte celular mediado por linfocitos T citotóxicos han involucrado el sistema de generación de poros celulares mediante la secreción de perforina/granzima en el microambiente del hepatocito infectado por virus. Sin embargo, se ha demostrado que los hepatocitos son resistentes a la acción citolítica de granzima B lo que sugiere que los linfocitos T citotóxicos inducen de forma predominante la muerte celular por apoptosis a través de Fas en hepatocitos infectados por virus. El TNF-a también parece jugar un papel muy relevante en la muerte celular por apoptosis de hepatocitos infectados por HCV y HBV. En este sentido, se ha detectado un incremento de la expresión de los receptores al TNF-R1 en hígado infectado, mayor producción de TNF-a en células mononucleares procedentes de pacientes infectados con HCV y HBV; así como un incremento de la susceptibilidad de la apoptosis dependiente de TNF-a inducida por proteínas virales en hepatocitos (15).
A pesar de la activación de la muerte celular por linfocitos T citotóxicos a través de Fas o por activación del receptor al TNF-a se han detectado mecanismos anti-apoptóticos inducidos por antígenos virales que pueden comportar tolerancia a la infección. En este sentido, la proteínas virales de HBV o HCV son capaces de estimular diversas vías intracelulares anti-apoptóticas relacionadas con la activación de factor nuclear kB (NF-kB) o JNK inducida por Fas (16,17) o por TNF-a (18) en hepatocitos. La inducción de mecanismos de resistencia a la muerte celular de los hepatocitos infectados es de una gran trascendencia para la persistencia de la infección, establecimiento de la lesión hepática crónica y desarrollo de HCC.
Carcinoma hepatocelular
El HCC es un tumor primario de hígado de elevada incidencia en la población. La patogenia del HCC es multifactorial, con una elevada asociación con la presencia de hepatitis viral crónica, consumo de alcohol, exposición a toxinas hepáticas, así como alteraciones genéticas como la hemocromatosis o deficiencia a a-antitripsina. La inducción de muerte celular es consecuencia de la respuesta de la célula afectada que comporta su eliminación del organismo y la no propagación de la modificación a las células circundantes. Por el contrario, la progresión del HCC es consecuencia de la alteración de los mecanismos pro-apoptóticos celulares que permiten la proliferación de la célula y propagación de las características de la célula modificada. En particular, las células tumorales presentan frecuentemente alterados los genes supresores de tumor, reparadores del DNA, reguladores del ciclo celular, así como los genes involucrados en la apoptosis celular.
Entre las alteraciones más frecuentemente detectadas en el HCC que afectan a la muerte celular están las mutaciones en el gen p53. La proteína p53 es el producto de un gen relacionado con la supresión de tumores que se activa como consecuencia de la lesión en el DNA celular. Tras la detección de la alteración génica, la proteína p53 induce la detención del ciclo celular para permitir la reparación del gen afectado. En caso de detectarse múltiples modificaciones génicas que requieren la eliminación de la célula mutada, p53 induce apoptosis gracias al incremento de expresión de proteínas pro-apoptóticas como Noxa, Puma, Bid o Bax, o bien induciendo la expresión de receptores involucrados en la muerte celular como Fas o TRAIL. En consecuencia, la alteración de p53 permite la supervivencia de la célula tumoral y la progresión del HCC (19). Otra alteración que se detecta frecuentemente en el HCC es la disminución de la expresión de Fas en las células tumorales que les permite escapar de la acción citolítica de los linfocitos T citotóxicos o células natural killer. Asimismo, la pérdida de la expresión del receptor Fas del HCC viene generalmente acompañada de la expresión excepcional del ligando de Fas en el hepatocito, generalmente restringida a células inflamatorias, que le permite inducir la muerte de las células del sistema inmune creando una zona privilegiada desde el punto de vista inmunológico. La reducción de la expresión de receptores Fas en HCC está negativamente correlacionado con su grado de diferenciación y con la supervivencia del paciente (20). Otros HCC presentan una elevada expresión de proteínas de la familia de Bcl-2 con actividad anti-apoptótica que confieren resistencia a los mecanismos intracelulares de inducción de apoptosis a través de la mitocondria. La inhibición de los procesos de muerte celular bien por alteración de los sistemas de detección y reparación del DNA, inductores de la muerte celular, alteración del ciclo celular o incremento de factores antiapoptóticos facilitan la estabilización de la lesión hepatocelular y progresión del HCC.
Bibliografía
1. Lemasters JJ. Mechanisms of hepatic toxicity. V. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Am J Physiol 1999; 276: G1-6. [ Links ]
2. Lemasters JJ. Dying a thousand deaths; redundant pathways from different organelles to apoptosis and necrosis. Gastroenterology 2005; 129: 351-60.
3. Cai J, Jones D. Superoxide in apoptosis: Mitochondrial generation triggered by cytochrome c loss. J Biol Chem 1998; 273: 401-4.
4. Morales A, Colell A, Marí M, et al. Glycosphingolipids and mitochondria: role in apoptosis and disease. Glyconjugate J 2004; 20: 579-88.
5. Bradham CA, Qian T, Streetz K, Trautwein C, Brenner DA, Lemasters JJ. The mitochondrial permeability transition is required for TNF-a -mediated apoptosis and cytochrome c release. Mol Cell Biol 1998; 18: 6353-64.
6. Aw TY, Andersson BS, Jones DP. Suppression of mitochondrial respiratory function after short-term anoxia. Am J Physiol 1987; 252: C362-8.
7. Marotto ME, Thurman RG, Lemasters JJ. Early midzonal cell death during low flow hypoxia in the isolated perfused rat liver: Protection by allopurinol. Hepatology 1988; 8: 585-90.
8. You M, Crabb DW. Molecular mechanisms of alcoholic fatty liver: role of sterol regulatory element-binding proteins. Alcohol 2004; 34: 39-43.
9. Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004; 114: 147-52.
10. Colell A, García-Ruiz C, Miranda A, et al. Selective glutathione depletion of mitochondria by ethanol sensitizes hepatocytes to tumor necrosis factor. Gastroenterology 1998; 115: 1541-51.
11. Pérez-Carreras M, Del Hoyo P, Martín MA, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003; 38: 999-1007.
12. Spivey JR, Bronk SF, Gores GJ. Glycochenodeoxycholate-induced lethal cell injury in rat hepatocytes. J Clin Invest 1993; 92: 17-24.
13. Hiramatsu N, Hayashi N, Katayama K, et al. Immunohistochemical detection of Fas antigen in liver tissue of patients with chronic hepatitis C. Hepatology 1994; 19: 1354-9.
14. Mochizuki K, Hayashi N, Hiramatsu N, et al. Fas antigen expression in liver tissue of patients with chronic hepatitis B. J Hepatol 1996; 24: 1-7.
15. Yoon JH, Gores GJ. Death receptors-mediated apoptosis and the liver. J Hepatol 2002; 37: 400-10.
16. Diao J, Khine AA, Sarangi F, Hsu E, Iorio C, Tibbles LA, et al. X protein of hepatitis B virus inhibits Fas-mediated apoptosis and is associated with up-regulation of the SAPK/JNK pathway. J Biol Chem 2001; 276: 8328-40.
17. Machida K, Tsukiyama-Kohara K, Seike E, Tone S, Shibasaki F, Shimizu M, et al. Inhibition of cytochrome c release in Fas-mediated signalling pathway in transgenic mice induced to express hepatitis C viral proteins. J Biol Chem 2001; 276: 12140-6.
18. Kim WH, Hong F, Jaruga B, Hu Z, Fan S, Liang TJ, et al. Additive activation of hepatic NF-kappa B by ethanol and hepatitis B protein X (HBX) or HCV core protein: Involvement of TNF-alpha receptor 1-independent and -dependent mechanisms. FASEB J 2001; 15: 2551-3.
19. Rocken C, Carl-McGrath S. Pathology and patohogenesis of hepatocellular carcinoma. Dig Dis 2001; 19: 269-78.
20. Nagao M, Nakajima Y, Hisanaga M, et al. The alterations of Fas receptor and ligand system in hepatocellular carcinomas: how do hematoma cells escape from the host immune surveillance in vivo? Hepatology 1999; 30: 413-21.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Jordi Muntané.
Unidad de Investigación.
Hospital Universitario Reina Sofía.
Avda. Menéndez Pidal, s/n.
E-14004 Córdoba.
e-mail: jordi.muntane.exts@juntadeandalucia.es
Recibido: 09-02-07.
Aceptado: 16-02-07.

