










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.99 no.12 Madrid dic. 2007
Profilaxis preprimaria de la hemorragia por varices
Pre-primary prophylaxis of variceal bleeding
R. González-Alonso, E. Garrido Gómez y A. Albillos Martínez
Servicio de Gastroenterología. Hospital Universitario Ramón y Cajal. Madrid
Dirección para correspondencia
RESUMEN
La formación de colaterales portosistémicas, en especial en la unión esofagogástrica, es una de las consecuencias más graves de la hipertensión portal. El aumento de la presión portal es la fuerza más importante que dirige la formación de varices esofagogástricas, siendo necesario para que esto ocurra que la presión portal (estimada por el gradiente de presión venosa hepática) alcance un valor mínimo de 10 mmHg. Posteriormente, la hiperemia esplácnica también contribuye al desarrollo de las varices. Las colaterales portosistémicas se forman por repermeabilización de vasos preexistentes, remodelado vascular y angiogénesis. El objetivo de la profilaxis preprimaria es evitar o retrasar la formación de varices esofagogástricas. En modelos experimentales de hipertensión portal, la administración precoz de vasoconstrictores esplácnicos como los beta-bloqueantes, de inhibidores de la síntesis de óxido nítrico o de sustancias anti-angiogénicas, inhibe la formación de colaterales portosistémicas. Sin embargo, los ensayos clínicos con beta-bloqueantes realizados en pacientes con cirrosis sin varices con objeto de retrasar su formación no han alcanzado los resultados esperados.
Palabras clave: Hipertensión portal. Varices esofágicas. Hemorragia digestiva alta. Profilaxis. Cirrosis hepática.
ABSTRACT
Portosystemic collateral formation, particularly at the gastroesophageal junction, is a most serious consequence of portal hypertension. Increased portal pressure is the most significant force underlying gastroesophageal variceal formation, to which end portal pressure (estimated from the hepatic venous pressure gradient) must reach at least 10 mmHg. Subsequently, splanchnic hyperemia also contributes to variceal development. Portoystemic collaterals result from repermeabilization of pre-extant vessels, vascular remodeling, and angiogenesis. The goal of pre-primary prophylaxis is preventing or delaying the formation of gastroesophageal varices. In experimental models of portal hypertension, early administration of splanchnic vasoconstrictors such as beta-blockers, nitric oxide synthesis inhibitors, or antiangiogenic substances inhibits portosystemic collateral formation. However, clinical trials of beta-blockers in patients with cirrhosis and no varices to delay variceal formation have failed to yield expected results.
Key words: Portal hypertension. Esophageal varices. Gastrointestinal bleeding. Prophylaxis. Cirrhosis of the liver.
Introducción
Las varices esofágicas son una manifestación clínica de la hipertensión portal, cuya causa más frecuente en nuestro medio es la cirrosis. La hemorragia digestiva alta por rotura de varices esofágicas es su complicación más grave, con una mortalidad elevada y una reducción de la supervivencia del 50% al año (1). El tratamiento actual de la hipertensión portal consiste en identificar a los pacientes que presentan varices, para administrar fármacos que al reducir la presión portal, también disminuyan la presión y la tensión variceal y con ello el riesgo de rotura y hemorragia. Otra estrategia que se ha planteado es identificar a los enfermos con presión portal elevada, pero que aún no han desarrollado varices, con objeto de tratarlos con fármacos reductores de la presión portal y así evitar que las varices lleguen a formarse. Este artículo describe las bases fisiopatológicas en las que se fundamenta esta estrategia, los estudios experimentales preclínicos que la sustentan y los ensayos clínicos realizados.
Relación entre el valor de la presión portal y el desarrollo de complicaciones
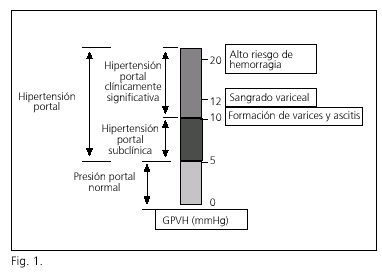
La presión portal es el resultado del producto del flujo venoso portal y de la resistencia a dicho flujo, y en la práctica clínica se estima indirectamente por el cálculo del gradiente de presión venosa hepática (GPVH), que corresponde a la diferencia entre la presión venosa en el sistema porta y en la circulación sistémica. En condiciones normales, el GPVH no supera los 5 mmHg; cuando excede dicho valor se considera que existe hipertensión portal. Existe una relación estrecha entre el valor del GPVH y el desarrollo de complicaciones derivadas del aumento de la presión portal. En concreto, el GPVH ha de alcanzar unos valores mínimos para que se formen y se rompan las varices y aparezca ascitis. Por extensión de estos datos, si se consigue reducir la presión portal por debajo de los valores umbrales es posible evitar la aparición de las complicaciones. Cuando el GPVH alcanza valores entre 5 y 10 mmHg existe hipertensión portal, pero esta aún no alcanza valores clínicamente significativos. La ascitis y las varices esofagogástricas no se forman hasta que el GPVH no iguala o supera los 10 mmHg, estado definido como "hipertensión portal clínicamente significativa" (2,3). Sólo cuando el GPVH alcanza los 12 mmHg puede aparecer la hemorragia digestiva alta por rotura de las mismas. Aquellos pacientes con un GPVH superior a 20 mmHg en las primeras 48 horas de la hemorragia presentan un riesgo 5 veces mayor de fracaso en el control de la hemorragia, de recidiva hemorrágica precoz y de muerte al año (4) (Fig. 1).
Fisiopatología de la hipertensión portal en la cirrosis
La hipertensión portal en la cirrosis es consecuencia de dos factores: el incremento de la resistencia intrahepática y el aumento del aflujo sanguíneo portal. Se ha demostrado en estudios experimentales que el factor que inicia la hipertensión portal es un aumento de la resistencia al flujo venoso portal. En la cirrosis este aumento de la resistencia ocurre a nivel de la microcirculación hepática, en el sinusoide, como consecuencia de dos mecanismos, uno mecánico, estructural, y otro funcional, dinámico, que es el responsable del 20-30% del incremento total de la resistencia. Los cambios estructurales en la arquitectura hepática, con fibrosis, nódulos de regeneración y capilarización de sinusoides, son los que determinan el mayor aumento en la resistencia vascular intrahepática (5), siendo casi todos ellos irreversibles. Algunas lesiones estructurales, particularmente el agrandamiento de los hepatocitos, pueden ser reversibles lentamente, conduciendo a la larga a una disminución de la presión portal, como ocurre en la hepatitis alcohólica con la abstinencia del alcohol (6).
Por su parte, el componente funcional, consecuencia de la contracción activa de los miofibroblastos portales/septales, de las células hepáticas estrelladas activadas y de las células musculares lisas de las vénulas portales, es reversible y puede ser modificado con tratamiento farmacológico (7). En el hígado sano, la zona de mayor resistencia vascular se sitúa a nivel del sinusoide hepático, que es capaz de contraerse y relajarse en respuesta a agentes vasoactivos. En condiciones normales, el aumento del tono vascular está modulado por un incremento de la actividad de los vasoconstrictores endógenos, como la endotelina, el estímulo alfa adrenérgico, los leucotrienos, el tromboxano A2 y la angiotensina A2, entre otros (8,9), y por vasodilatadores endógenos, como el óxido nítrico (ON) y las prostaciclinas. En la cirrosis, existe un desequilibrio entre los agentes vasoconstrictores y los vasodilatadores que va a determinar un aumento de la resistencia vascular intrahepática: las sustancias vasoconstrictoras están aumentadas, mientras que la producción intrahepática de ON está disminuida (10,11). El factor más importante responsable del aumento de la resistencia vascular del hígado cirrótico es un déficit en la síntesis endotelial de ON, que impide que se contrarresten los estímulos vasoconstrictores, que a su vez, están aumentados en la cirrosis. Por otro lado, se ha demostrado que en hígados normales aumentan la expresión de la enzima sintetasa del ON endotelial (eONS) y la liberación de ON, en respuesta al estrés de rozamiento que causa en el endotelio sinusoidal un aumento del flujo sanguíneo (12). Este incremento en la síntesis y liberación de ON, permite que se dilate el sinusoide y la presión en su interior no aumente cuando lo hace el flujo sanguíneo, por ejemplo, después de la ingesta. Sin embargo, en hígados con cirrosis el ON no sólo está disminuido en condiciones basales, sino que no aumenta de manera apropiada en respuesta a dicho estrés, lo que conlleva una alteración del flujo y un incremento en la resistencia vascular intrahepática. Se ha observado en ratas con cirrosis y ascitis una "vasoconstricción paradójica" en respuesta a agonistas de la eONS, como es la acetilcolina, con una disminución de la vasorrelajación, que se atribuye a la pérdida del efecto vasodilatador del ON derivado de endotelio (10). Este déficit intrahepático de ON se produce a pesar de niveles normales de la eONS, reflejando una alteración a nivel de los mecanismos reguladores post-traduccionales de la actividad enzimática. La eONS está sometida a una importante regulación post-traduccional, por diferentes factores activadores, como la calmodulina, e inhibidores, como la caveo-lina-1, de su capacidad de síntesis de ON (12,13).
Pero más allá de la pérdida del efecto vasodilatador, el déficit de ON va a inducir un aumento de la resistencia intrahepática a través de otros mecanismos: trombogénesis, fibrogénesis, inflamación y aumento del estrés oxidativo (11). El ON actúa en fases tempranas de la aterogénesis, inhibiendo la agregación, adhesión y activación plaquetarias que aparecen estimuladas por el estrés endotelial presentes en la microcirculación hepática de la cirrosis. De esta manera, una disminución de la biodisponibilidad del ON va a acompañarse de un aumento en la trombogénesis, con formación de trombos a nivel de las ramas de las venas porta y hepática (14). Además, el ON posee propiedades antiinflamatorias inhibiendo la expresión de proteínas de adhesión leucocitaria, protegiendo al endotelio del estrés oxidativo e inhibiendo la peroxidación lipídica (15). Por último, el déficit de ON que existe en la cirrosis puede estar involucrado en la síntesis y depósito de colágeno en el espacio de Disse (16), ya que el ON en condiciones normales disminuye la síntesis de colágeno por las células musculares lisas y actúa directamente en la generación de la matriz extracelular. La suma de todos estos efectos inducidos por la disminución intrahepática de ON va a contribuir de manera significativa al aumento de la resistencia vascular intrahepática y al progreso de la hipertensión portal. La demostración de la existencia de un déficit de ON a nivel hepático constituye la base racional para el uso de nitratos orgánicos, como el 5-mononitrato de isosorbida (17), y de otras estrategias que aumentan la producción de ON a nivel intrahepático, como los donantes de ON selectivos hepáticos, entre ellos el NCX-1000 (18), y los fármacos que actúan sobre la regulación post-traduccional de la eONS, como las estatinas (19,20).
El segundo factor que contribuye a aumentar la presión portal en la cirrosis es el aumento del aflujo venoso portal, el cual es secundario a la vasodilatación arteriolar esplácnica, y es característico de estadios más avanzados de cirrosis (21). El aumento del aflujo de sangre al territorio venoso portal mantiene y agrava la hipertensión portal establecida, y explica por qué persiste a pesar de formarse las colaterales portosistémicas. Este hiperaflujo es consecuencia del incremento en la concentración de vasodilatadores circulantes, de la excesiva producción local de vasodilatadores endoteliales y de una respuesta vascular deprimida a los vasoconstrictrores endógenos. Entre los vasodilatadores de origen endotelial el más estudiado ha sido el ON. En contra de lo que ocurre en la microcirculación intrahepática, en las vasculaturas esplácnica y sistémica existe una sobreproducción endotelial de ON (22). Este incremento del ON es el principal mediador de la vasodilatación arterial y del desarrollo de la circulación hiperdinámica. Se ha demostrado que un aumento en la producción de ON en el territorio vascular esplácnico de ratas con hipertensión portal es el responsable de la "hiporrespuesta" a vasoconstrictores (23). La inhibición de la síntesis de ON en ratas cirróticas previene, aunque no completamente, el desarrollo de la circulación hiperdinámica, con una normalización de la presión arterial, del gasto cardiaco y de las resistencias vasculares sistémicas, y además, disminuye la colateralización portosistémica (24,25).
Varios estudios en hipertensión portal han observado que el aumento del ON está mediado principalmente por la eONS (26,27). Los factores que estimulan la actividad de la eONS son el estrés sobre la pared del vaso que causa el propio flujo hiperdinámico, la traslocación bacteriana y la colateralización portosistémica (11,22). Las endotoxinas bacterianas activan las células mononucleares, las cuales producen citoquinas proinflamatorias, como el TNF-alfa, que estimula la producción de ON al aumentar la actividad de la eONS (27-29). El aumento de la actividad de la eONS precede a la aparición de los tres factores mencionados, por lo que debe existir un estímulo del enzima en las fases iniciales de la hipertensión portal (30). En este sentido, se ha constatado en ratas con hipertensión portal prehepática que la vasoconstricción arterial mesentérica que se produce tras la inducción de hipertensión portal, y que precede a la hiperemia esplácnica podría ser el estímulo inicial de la eONS (31).
El ON no es el único agente implicado en la patogenia de la hiperemia esplácnica de la hipertensión portal, habiéndose demostrado también la participación del glucagón y de productos de la ciclooxigenasa. Es importante señalar que la patogenia del estado circulatorio hiperdinámico de la hipertensión portal no puede adscribirse a un único vasodilatador. De hecho, la hiperemia esplácnica también se desarrolla cuando se induce hipertensión portal en modelos experimentales genéticamente deficientes de ON (30), o a los que se inhibe crónicamente la síntesis de ON o la ciclooxigenasa (29,32,33). En estos estudios experimentales en los que se inhibe la síntesis o la acción de uno de los vasodilatadores potencialmente implicados, la hiperemia esplácnica se desarrolla por el aumento compensador de los otros.
Desarrollo de la circulación colateral
Las colaterales portosistémicas, entre ellas las varices esofagogástricas, se desarrollan en respuesta al aumento de la presión portal, como un mecanismo para descomprimir la vena porta y permitir el retorno venoso a la circulación sistémica. A pesar de que la mayor parte del flujo venoso se desvía por estas colaterales, la hipertensión portal se mantiene a expensas de un aumento concomitante del aflujo venoso portal. De esta forma, las colaterales portosistémicas son las responsables de las complicaciones más graves de la hipertensión portal, como la hemorragia por varices y la encefalopatía hepática.
Los estímulos que promueven el proceso de colateralización en la hipertensión portal son el aumento de la presión y del aflujo venoso portal. El incremento de la presión portal es el factor más importante, pues las colaterales sólo se forman cuando el GPVH es mayor de 10 mmHg (2,34). El riesgo de hemorragia variceal disminuye cuando se reduce el GPVH, indicando que la reducción de la presión portal es la diana más efectiva para impedir la formación y/o el desarrollo de vasos colaterales. Sin embargo, aunque en modelos experimentales de cirrosis se ha podido prevenir la formación de colaterales al administrar fármacos que disminuyen la presión portal, como el propanolol (35), ello no ha sido así en enfermos con cirrosis compensada tratados con betabloqueantes no selectivos (36). La hiperemia esplácnica y el aumento del aflujo venoso portal es la segunda fuerza promotora del desarrollo de colaterales. Hipertensión portal e hiperemia esplácnica son dos factores relacionados, pero independientes, en la formación y desarrollo de la circulación porto-colateral (24,37).
Los mecanismos que modulan el desarrollo de las colaterales portosistémicas son múltiples. La colateralización se produce tanto por apertura de vasos preexistentes como por formación de vasos nuevos (24,38). El primero de estos mecanismos es una respuesta adaptativa a cambios crónicos en el flujo sanguíneo. Incrementos persistentes del flujo desencadenan señales endoteliales que modifican la estructura del vaso, aumentando su diámetro y con ello su capacidad para recibir un mayor flujo (39). El otro mecanismo implicado en la colateralización portosistémica de la hipertensión portal es la angiogénesis. Esta es la formación de nuevos vasos mediada por la proliferación de células endoteliales in situ y de células del músculo liso, cuyos progenitores se reclutan de la sangre (38). De nuevo, el ON desempeña un papel muy importante en ambos procesos, remodelado vascular con expansión luminar y angiogénesis (40,41). El desarrollo de colaterales y el hiperaflujo portocolateral provocan un aumento del estrés de la pared del vaso, que es un potente estímulo para la síntesis de ON, el cual promueve la angiogénesis y la dilatación de los vasos (11). Se ha demostrado que la inhibición de la síntesis de ON en modelos experimentales de hipertensión portal antagoniza la respuesta angiogénica, reduce la hiperemia esplácnica y el grado de colateralización portosistémica y atenúa el remodelado vascular (24,38,42).
El proceso de la angiogénesis está mediado de forma primaria por el factor de crecimiento endotelial vascular (VEGF) (43). Se ha observado que la administración crónica de inhibidores de los receptores de la VEGF a ratas con hipertensión portal previene la formación de colaterales portosistémicas (44). Diversos mecanismos, implicados a su vez en la patogénesis de la hipertensión portal, como la hipoxia tisular, factores de crecimiento, como el factor de crecimiento de fibroblastos (FGF), algunas citoquinas proinflamatorias, como las interleuquinas 1 y 6, y el estrés de rozamiento, van a inducir la expresión del gen VEGF y promueven la angiogénesis en diversas células (45). Además, parece existir una interrelación entre la producción de ON y de VEGF. El ON media los efectos endoteliales del VEGF, facilitando la proliferación de células endoteliales y la migración, y también estimula la liberación de progenitores de células endoteliales de la médula ósea, contribuyendo a la vasculogénesis (46). Por su parte, el VEGF promueve la producción de ON e induce la expresión de la eONS (47); experimentalmente se ha demostrado que un aumento leve de la presión portal estimula la producción de VEGF en la microcirculación yeyunal, y esta señal es suficiente para inducir la actividad de la eONS (48). Este conjunto de datos indica que tanto la eONS como el VEGF son dianas potenciales para modular la formación de colaterales portosistémicas (varices) en la hipertensión portal.
Modulación de la formación de colaterales portosistémicas en modelos experimentales de HTP: estudios preclínicos
Los primeros intentos de modificar la formación de colaterales portosistémicas se llevaron a cabo en modelos de hipertensión portal, prehepática (ratas con ligadura parcial de vena porta) e intrahepática (esquistosomiasis murina, cirrosis inducida por tetracloruro de carbono) (Tabla I). En estos modelos experimentales se comprobó que era posible reducir la formación de colaterales portosistémicas causando vasoconstricción arteriolar y atenuación de la hiperemia esplácnica, como factores responsables de la colateralización portosistémica. Ello puede lograrse administrando bloqueantes beta-adrenérgicos no selectivos (35,49,50), como el propanolol, o inhibidores de la síntesis de ON, como los análogos de la L-arginina (24), o anticuerpos monoclonales frente al TNF-alfa (29). Sarin y cols. fueron los primeros en demostrar que la administración crónica de propranolol atenuaba el desarrollo de colaterales portosistémicas en un modelo murino de hipertensión portal por esquistosomiasis (35), hecho después corroborado en ratas con cirrosis inducida por tetracloruro de carbono (49,50). El propranolol reduce el flujo portocolateral por disminución del aflujo venoso portal, secundario a vasoconstricción esplácnica, y de la presión portal (52,53), factores implicados en la formación y desarrollo de vasos colaterales. Posteriormente, se demostró que la reducción de la síntesis de ON, mediante inhibidores de la eONS, como la N-Nitro-L-arginina (24), atenuaba la formación de vasos colaterales en ratas con hipertensión portal prehepática. Este efecto se cree que es secundario a la reducción de la hiperemia esplácnica y del aflujo venoso portal que causa la inhibición de la síntesis de ON, pues la presión portal no se modifica. Hecho que no hace sino enfatizar la relevancia del aumento de flujo esplácnico en el desarrollo de la colateralización portosistémica. Consecuencias similares se logran inhibiendo el TNF-alfa circulante, citoquina proinflamatoria cuya concentración sérica está aumentada en la hipertensión portal, y que estimula la actividad de la eONS (29). Más recientemente, se ha demostrado que el bloqueo de mediadores específicos de la angiogénesis, como el VEGF, también atenúa la colateralización portosistémica (44,51). Fernández y cols. observaron que el desarrollo de los vasos portocolaterales en ratas con hipertensión portal se asociaba con la presencia de angiogénesis en el territorio esplácnico, evidenciado por un incremento cronológico y significativo de la expresión del VEGF, del receptor del VEGF y de la proteína endotelial CD31, relacionada con la vasculogénesis y la angiogénsis. La administración de anticuerpos monoclonales frente al receptor VEGF disminuía la neovascularización intestinal que existía en estos animales, y ello ocurría a pesar de mantenerse elevada la presión portal.
En suma, estos estudios experimentales han demostrado que es posible modular el grado de colateralización portosistémica en la hipertensión portal, han identificado potenciales dianas terapéuticas para ello y han aportado la base racional para la traslación clínica de estos hallazgos.
Profilaxis preprimaria de las varices esofágicas
La hipertensión portal es una entidad clínica dinámica y progresiva en cuyo curso se desarrolla gradualmente la circulación colateral, incluyendo las varices esofágicas. Se sabe por estudios longitudinales que las varices se desarrollarán en todos los enfermos con cirrosis e hipertensión portal, y una vez que aparecen tienden a crecer en tamaño y sangrar (54). La prevalencia de varices esofágicas es proporcional a la gravedad de la enfermedad hepática y en el momento del diagnóstico de la cirrosis es aproximadamente del 40% en los pacientes con cirrosis compensada y del 60% en aquellos con ascitis. Las varices se forman a un ritmo anual del 5% y sólo cuando el GPVH es mayor de 10 mmHg. Aunque el riesgo de hemorragia es bajo en pacientes sin varices (1-2% al año) o con varices pequeñas (5% al año), las varices pequeñas tienen una tasa de crecimiento a varices medianas o grandes del 10 al 15% al año, siendo el ritmo mayor en los pacientes con cirrosis de etiología etílica o avanzada (Child-Pugh B-C). Por su parte, las varices grandes tienen un riesgo anual de sangrado del 15% (55-57). El riesgo medio de presentar al menos un episodio de sangrado en los enfermos con cirrosis y ascitis es del 30%, con el mayor riesgo para aquellos con varices grandes y enfermedad descompensada. Por otra parte, un episodio aislado de hemorragia variceal presenta un riesgo de muerte precoz del 5-8%, y una mortalidad a las 6 semanas del 20-30%, y además, la probabilidad de supervivencia al año se reduce al 50% (1,58). Estos datos explican la importancia clínica de la profilaxis de la hemorragia digestiva por varices.
La profilaxis preprimaria tiene como objetivo evitar o retrasar la formación de varices esofágicas, así como prevenir su crecimiento cuando ya se han formado y son aún de pequeño tamaño. Los únicos fármacos que se han utilizado para profilaxis preprimaria en estudios clínicos son los beta-bloqueantes no cardioselectivos, que han demostrado claramente su utilidad en la profilaxis primaria de la hemorragia por varices (59). El mecanismo de acción de los betabloqueantes es complejo y varía de acuerdo a la dosis. Bajas dosis provocan vasoconstricción esplácnica a través del bloqueo de los receptores adrenérgicos beta2, lo que conduce a una disminución del flujo sanguíneo portal y, por consiguiente, a una reducción de la presión portal. Al aumentar la dosis, el efecto negativo sobre el gasto cardiaco, a través del bloqueo de los receptores adrenérgicos beta1, va a disminuir el flujo portal. Los primeros estudios que se realizaron con betabloqueantes en la profilaxis preprimaria fueron en modelos animales de cirrosis e hipertensión portal, donde se observó que la administración precoz de estos fármacos en la historia natural de esta enfermedad era capaz de reducir el desarrollo de la circulación colateral (35,49-51). Los betabloqueantes al disminuir el flujo sanguíneo hacia el sistema esplácnico, reducen el estímulo para la formación de colaterales portosistémicas. Por otra parte, en estudios fisiopatológicos en humanos también se ha demostrado que el efecto de los betabloqueantes para disminuir la presión portal es más evidente en enfermos con cirrosis en estadios iniciales que presentan un menor desarrollo de la circulación colateral (60). Estos hallazgos han constituido la base racional para el diseño de estudios clínicos destinados a testar la hipótesis de que la administración precoz de betabloqueantes puede prevenir la formación de varices esofágicas.
Hasta el momento hay publicados en la literatura 3 estudios multicéntricos, controlados y aleatorizados, con betabloqueantes para prevenir la formación, desarrollo y/o crecimiento de las varices esofágicas en enfermos con cirrosis e hipertensión portal (36,61,62) (Tabla II). Todos ellos utilizan un beta bloqueante frente a placebo, pero el tipo de betabloqueante es diferente en cada uno de los estudios. En el más antiguo, Calès y cols. (61) trataron 206 pacientes con cirrosis y sin varices o con varices pequeñas con propanolol a dosis fijas, y les siguieron durante un tiempo medio de 2 años, sin demostrar un efecto beneficioso de los betabloqueantes frente a placebo en la incidencia de varices grandes ni en el riesgo de hemorragia. Merkel y cols. (62) trataron 161 pacientes con cirrosis y varices pequeñas con nadolol. Tras 5 años de seguimiento medio se demostró que los pacientes tratados con nadolol tenían menor riesgo de crecimiento y mayor tasa de regresión de las varices, con una incidencia menor de hemorragia. El tercer estudio, realizado por Groszmann y cols. (36), incluyó 213 enfermos con cirrosis y sin varices, tratados con timolol y seguidos una media de 4,6 años. En este caso, al igual que en el primero, tampoco se observó un efecto beneficioso del betabloqueante, de forma que la incidencia de formación de varices y de hemorragia fueron similares en el grupo placebo y en el grupo de timolol. Además, la tasa de efectos adversos graves fue mayor en el grupo del betabloqueante.
La disparidad de resultados en los tres estudios puede ser explicada en parte por heterogeneidad clínica. Por un lado, existe heterogeneidad en el tipo de enfermos incluidos. El estudio de Merkel y cols. sólo incluyó pacientes con varices esofágicas pequeñas, el de Groszmann y cols. sólo cirrosis con hipertensión portal y sin varices, y en cambio en el de Calès y cols., se incluyeron los 2 tipos de enfermos. Se sabe que entre los enfermos con cirrosis y sin varices existen 2 tipos clínicos, aquellos con hipertensión portal subclínica en los que el grado de aumento de la presión portal no es suficiente para que se formen las varices, y que representa el mayor porcentaje de estos pacientes, y aquellos con hipertensión portal clínicamente significativa con desarrollo de la circulación colateral y en los que se podrían desarrollar varices a nivel esofágico. Como demuestra el estudio de Groszmann y cols., los betabloqueantes puede que no tengan utilidad en pacientes cuyo GPVH es menor de 10 mmHg. Por otra parte, el tipo de betabloqueante y la dosis utilizada no fueron iguales en los tres estudios. En el de Calès y cols. se administró una dosis fija de propranolol que pudo sobre o infratratar determinados tipos de enfermos; y en el de Groszmann y cols. no se pudo alcanzar en todos los pacientes una dosis óptima de timolol capaz de reducir el GPVH, por la aparición de efectos adversos. Además, el mecanismo de acción de estos fármacos no es exactamente el mismo, y ello podría influir en el grado de disminución de la presión portal. Otra fuente de heterogeneidad y de confusión es el porcentaje de alcohólicos y de abstinencia durante los estudios; se sabe que la abstinencia de alcohol puede por sí misma retrasar el crecimiento y progresión de las varices, incluso su regresión, al provocar una disminución en el GPVH, lo que podría enmascarar el efecto del tratamiento.
Por el momento y con la evidencia científica disponible no se puede indicar la administración de betablo-queantes a pacientes con cirrosis e hipertensión portal, pero sin varices esofágicas con objeto de impedir la formación de estas y las complicaciones asociadas. Es posible que estos fármacos tengan menor efecto en las fases tempranas de la cirrosis. El diseño de ensayos clínicos en estadios precoces de cirrosis con fármacos diferentes a los betabloqueantes y que actúen a nivel de otros mecanismos fisiopatológicos, podría tener un efecto más beneficioso en la prevención del desarrollo y progresión de la circulación colateral, y en concreto, de las varices esofágicas.
Bibliografía
1. Bosch J, García Pagán JC. Prevention of variceal rebleeding. Lancet 2003; 361: 952-4. [ Links ]
2. García Tsao G, Groszmann RJ, Fisher RL, Conn HO, Atterbury CE, Glickman M. Portal pressure, presence of gastroesophageal varices and variceal bleeding. Hepatol 1985; 5: 419-24. [ Links ]
3. Groszmann RJ, Glickman M, Blei AT, Storer E, Conn HO. Wedged and free hepatic venous pressure measured with a balloon catheter. Gastroenterol 1979; 76: 253-8. [ Links ]
4. Moitinho E, Escorsell A, Bandi JC, Salmerón JM, García Pagán JC, Rodés J, et al. Prognostic value of early measurements of portal pressure in acute variceal bleeding. Gastroenterol 1999; 117: 626-31. [ Links ]
5. Popper H, Zalk FG. Pathological aspects of cirrhosis. Am J Med 1958; 1958: 24: 593-625. [ Links ]
6. Colman JC, Britton RS, Orrego H, Valdivia V, Medline A, Israel Y. Relation between osmotically induced hepatocyte enlargement and portal hypertension. Am J Physiol 1983; 245: G382-7. [ Links ]
7. Bhathal PS, Grossman HJ. Reduction of the increased portal vascular resistance of the isolated perfused cirrhotic rat liver by vasodilators. J Hepatol 1985; 1: 325-37. [ Links ]
8. Jockey DC, Weisger RA. Endothelin induced contractility of stellate cells from normal and cirrhotic rat liver: Implications for regulation of portal pressure and resistance. Hepatol 1996; 24: 233-40. [ Links ]
9. Ballet F, Chretien Y, Rey C, Poupon R. Differential response of normal and cirrhotic livers to vasoactive agents. A study in the isolated perfused rat liver. J Pharmacol Exp Ther 1988; 244: 233-5. [ Links ]
10. Gupta TK, Toruner M, Chung MK, Groszmann RJ. Endothelial dysfunction and decreased production of nitric oxide in the intrahepatic microcirculation effects on portal hemodynamics and on liver and renal function. Hepatol 1998; 28: 926-31. [ Links ]
11. Wiest R, Groszmann RJ. The paradox of nitric oxide in cirrhosis and portal hypertension: too much, not enough. Hepatol 2002; 35: 478-91. [ Links ]
12. Shah V, Toruner M, Haddad F, Cadelina G, Papapetropoulos A, Choo K, et al. Impaired endothelial nitric oxide synthase activity associated with enhanced caveolin binding in experimental cirrhosis in the rat. Gastroenterology 1999; 117: 1222-8. [ Links ]
13. Nedvetsky PI, Sessa WC, Schmidt HH. There´s NO binding like NOS binding: protein-protein interactions in NO/cGMP signaling. Proc Natl Acad Sci USA 2002; 99: 16510-2. [ Links ]
14. Wanless IR, Wong F, Blendis LM, Greig P, Heathcote EJ, Levy G. Hepatic and portal vein thrombosis in cirrhosis: possible role in development of parenchymal extinction and portal hypertension. Hepatol 1995; 21: 1238-47. [ Links ]
15. Keaney J, Vita J. Atheroesclerosis, oxidative stress, and antioxidant protection in endhotelium-derived relaxing factor action. Prog Cardiovasc Dis 1995: 129-54. [ Links ]
16. Casini A, Ceni E, Salzano R, Biondi P, Parola M, Galli A, et al. Neutrophil-derived superoxide anion induces lipid peroxidation and stimulates collagen synthesis in human hepatic stellate cells: role of nitric oxide. Hepatol 1997; 25: 361-7. [ Links ]
17. García Pagán JC, Feu F, Navasa M, Bru C, Ruiz DA, Bosch J, et al. Long term haemodynamic effects of isosorbide 5-mononitrate in patients with cirrhosis and portal hypertension. J Hepatol 1990; 11 (2): 189-95. [ Links ]
18. Shah V, Chen AF, Cao S, Hendrickson H, Weiler D, Smith L, et al. Gene transfer of recombinant endothelial nitric oxide synthase to liver in vivo and in vitro. Am J Physiol Gastrointest Liver Physiol 2000; 279: G1023-G30. [ Links ]
19. Laufs U, La F, V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 1998; 97: 1129-35. [ Links ]
20. Zafra C, Abraldes J, Cortez C, Berzigotti A, Tarantino I, Gracía-Pagán JC, et al. Simvastatin ammeliorates the increased hepatic vascular tone in patients with cirrhosis. J Hepatology 2003; 38 (Supl. 2): 34. [ Links ]
21. Sikuler E, Groszmann RJ. Interaction of flow and resistance in maintenance of portal hypertension in a rat model. Am J Physiol 1986; 250: G205-G212. [ Links ]
22. Hori N, Wiest R, Groszmann RJ. Enhanced release of nitric oxide in response to changes in flow and shear stress in the superior mesenteric arteries of portal hypertensive rats. Hepatol 1998; 28: 1467-73. [ Links ]
23. Lee FY, Albillos A, Colombato LA, Groszmann RJ. The role of nitric oxide in the vascular hyporesponsiveness to methoxamine in portal hypertensive rats. Hepatol 1992; 16: 1043-8. [ Links ]
24. Lee FY, Colombato LA, Albillos A, Groszmann RJ. Administration of N omega nitro-L-arginine ameliorates portal-systemic shunting in portal hypertensive rats. Gastroenterol 1993; 105: 1464-70. [ Links ]
25. Pizcueta P, Pique JM, Fernández M, Bosch J, Rodes J, Whittle BJ, et al. Modulation of the hyperdinamic circulation of cirrhotic rats by nitric oxide inhibition. Gastroenterol 1992; 103: 1909-15. [ Links ]
26. Fernández M, García Pagán JC, Casadevall M, et al. Evidence against a role for inducible nitric oxide synthase in the hyperdinamic circulation of portal-hypertensive rats. Gastroenterol 1995; 108: 1487-95. [ Links ]
27. Wiest R, Das S, Cadelina G, García Tsao G, Milstien S, Groszmann RJ. Bacterial translocation in cirrhotic rats stimulates eNOS-derived NO productionand impairs mesenteric vascular contractility. J Clin Invest 1999; 104: 1223-33. [ Links ]
28. Chu C, Lee F, Wang S, Lu R, Tsai Y, Lin H, et al. Hyperdinamic circulation of cirrhotic rats with ascites: role of endotoxin, tumor necrosis factor and nitric oxide. Clinical Science 1997; 93: 219-25. [ Links ]
29. Muñoz J, Albillos A, Pérez Paramo M, Rossi I, Álvarez Mon M. Factors mediating the hemodynamic effects of tumor necrosi [ Links ]
30. Iwakiri Y, Cadelina G, Sessa WC, Groszmann RJ. Mice with targeted deletion of eNOS develop hyperdynamic circulation associated with portal hypertension. Am J Physiol Gastrointest Liver Physiol 2002; 283: G1074-81. [ Links ]
31. Tsai MH, Iwakiri Y, Cadelina G, et al. Mesenteric vasocontriction triggers nitric overproduction in the superior mesenteric artery of portal hypertensive rats. Gastroenterol 2003; 125: 1452-61. [ Links ]
32. García Pagán JC, Fernández M, Bernadich C, Pizcueta P, Pique JM, Bosch J, et al. Effects of continued NO inhibition on portal hypertensive syndrome after portal vein stenosis in rats. Am J Physiol 1994; 267: G984-990. [ Links ]
33. Fernández M, García Pagán JC, Casadevall M, Mourelle MI, Pique JM, Bosch J, et al. Acute and chronic cyclooygenase blockage in portal-hypertensive rats: influence in nitric oxide byosynthesis. Gastroenterology 1996; 110: 1529-35. [ Links ]
34. Viallet A, Marleau D, Huet M, et al. Hemodynamic evaluation of patients with intrahepatic portal hypertension. Gastroenterol 1975; 69: 1297-300. [ Links ]
35. Sarin S , Groszmann RJ, Mosca PG, et al. Propranolol ameliorates the development of portal-systemic shunting in a chronic murine schistosomiasis model of portal hypertension. J Clin Invest 1991; 87: 1032-6. [ Links ]
36. Groszmann RJ, García Tsao G, Bosch J, Grace N, Burroughs A, Planas R, et al. Beta-blockers to prevent gastroesophageal varices in patients with cirrhosis. N Engl J Med 2005; 353: 2254-61. [ Links ]
37. Albillos A, Banares R, González M, Catalina MV, Pastor O, González R, et al. The extent of the collateral circulation influences the postprandial increase in portal pressure in cirrhotic patient. Gut 2007; 56: 259-64 [ Links ]
38. Sumanovski LT, Battegay E, Stumm M, van der ooij M, Sieber CC. Increased angiogenesis in portal hypertensive rats: Role of nitric oxide. Hepatol 1999; 29: 1044-9. [ Links ]
39. Gibbons GH, Dzau VJ. The emerging concept of vascular remodeling. N Engl J Med 1994; 330: 1431-8. [ Links ]
40. Ziche M, Morbidelli L, Masini E, Amerini S, Granger H, Maggi C, et al. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vivo promoted by substance. J Clin Invest 1994; 94: 2036-44. [ Links ]
41. Rudic R, Shesely E, Maeda N, Smithies O, Segal S, Sessa W. Direct evidence for the importance of endothelium-derived nitric oxide in vascular remodelling. J Clin Invest 1998; 101: 731-6. [ Links ]
42. Fernández Varo G, Ros J, Morales Ruiz M, Cejudo Martín P, Arroyo V, Solé M, et al. Nitric oxide synthase 3-dependent vascular remodeling and circulatory dysfunction in cirrhosis. Am J Pathol 2003; 162: 1985-93. [ Links ]
43. Moreau R. VEGF-induced angiogenesis drives collateral circulation in portal hypertension. J Hepatol 2005; 43: 6-8. [ Links ]
44. Fernández M, Vizzutti F, García Pagán JC, Rodés J, Bosch J. Anti-VEGF receptor-2 monoclonal antibody prevents portal-systemic collateral vessel formation in portal hypertensive mice. Gastroenterolo 2004; 126: 886-94. [ Links ]
45. Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocrine Rev 1997; 18: 4-25. [ Links ]
46. Aicher A, Heeschen C, Mildner Rihm C, Urbich C, Ihling C, Technau Ihling, et al. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med 2003; 9: 1370-6. [ Links ]
47. Papapetropoulos A, García Cardena G, Madri JA, Sessa WC. Nitric oxide production contributes to the angiogenic properties of vascular endothelial growth factor in human endothelial cells. J Clin Invest 1997; 100: 3131-9. [ Links ]
48. Abraldes JG, Iwakiri Y, Loureiro Silva M, Haq O, Sessa WC, Groszmann RJ. Mild increases in portal pressure upregulate vascular endothelial growth factor and endothelial nitric oxide synthase in the in-testinal microcirculatory bed, leading to a hyperdynamic state. Am J Physiol Gastrointest Liver Physiol 2006; 290: G980-7. [ Links ]
49. Colombato LA, Albillos A, Genecin P, et al. Prevention of portal-systemic shunting in propranolol treated and in sodium-restricted cirrhosis rats. Gastroenterology 1991; 100: A730. [ Links ]
50. Lin HC, Soubrane O, Cailmail S, Lebrel D. Early chronic administration of propranolol reduces the severity of portal hypertension and portal-systemic shunts in conscious portal vein stenosed rats. J Hepatol 1991; 13: 213-9. [ Links ]
51. Fernández M, Mejias M, Angermayr B, García Pagán JC, Rodés J, Bosch J. Inhibition of VEGF receptor-2 decreases the development of hyperdinamic splachnic circulation and portal-systemic collateral vessels in portal hypertensive rats. J Hepatol 2005; 3: 98-103. [ Links ]
52. roeger R, Groszmann RJ. The effect of selective blockade of b2-adrenergic receptors on portal and systemic hemodynamics in a portal hypertensive rat model. Gastroenterology 1985; 88: 896-900. [ Links ]53. Bosch J, Mastai R, ravetz D, Bruix J, Gaya J, Rigau J, et al. Effects of propranolol on azygos blood flow and hepatic and systemic hemodynamics in cirrhosis. Hepatology 1984; 4: 1200-5. [ Links ]
54. Christiensen E, Fauerholdt L, Schlichting P, Juhl E, Poulsen H, Tygstrup N. Aspects of natural history of gastrointestinal bleeding in cirrhosis and the effect of prednisone. Gastroenterol 1981; 81: 944-52. [ Links ]
55. Zoli M, Merkel C, Magalotti D, Gueli C, Grimaldi M, Gatta A, et al. Natural history of cirrhotic patients with small esophageal varices: a prospective study. Am J Gastroenterol 2000; 95: 503-8. [ Links ]
56. Merli M, Nicolini G, Angeloni S, Rinaldi V, De SA, Merkel C, et al. Incidence and natural history of small esophageal varices in cirrhotic patients. J Hepatol 2003; 38: 266-72. [ Links ]
57. Calés P, Desmorat H, Vinel JP, Caucanas JP, Ravaud A, Gerin P, et al. Incidence of large oesophageal varices in patients with cirrhosis: Application to prophylaxis of first bleeding. Gut 1990; 31: 1298-302. [ Links ]
58. Graham DY, Smith JL. The course of patients after variceal hemorrhage. Gastroenterology 1981; 80: 800-9. [ Links ]
59. D´Amico G, Pagliaro L, Bosch J. Pharmacological treatment of portal hypertension: an evidence-based approach. Semin Liver Dis 1999; 19: 75-505. [ Links ]
60. Escorsell A, Ferayorni L, Bosch J, García Pagán JC, García Tsao G, Grace ND, et al. The portal pressure response to beta-blockade is greater in cirrhotic patients without varices than in those with varices. Gastroentrology 1997; 112: 2012-6. [ Links ]
61. Cales P, Oberti F, Payen JL, Naveau S, Guyader D, Blanc P, et al. Lack of effect of propranolol in the prevention of large oesophageal varices in patients with cirrhosis: A randomized trial. Eur J Gastroenterol Hepatol 1999; 11: 741-5. [ Links ]
62. Merkel C, Marín R, Angeli P, Zanella P, Felder M, Bernardinello E, et al. A placebo-controlled clinical trial of nadolol in the prophylaxis of growth of small esophageal varices in cirrhosis. Gastroenterol 2004 127: 476-8. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Agustín Albillos Martínez.
Servicio de Gastroenterología.
Hospital Universitario Ramón y Cajal.
Ctra. de Colmenar km 9,600.
2803 Madrid
e-mail: aalbillosm@meditex.es

