










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.104 no.3 Madrid mar. 2012
https://dx.doi.org/10.4321/S1130-01082012000300009
Multiple desmoid tumors in a patient with familial adenomatous polyposis caused by the novel W421X mutation
Tumor desmoide múltiple en un paciente con poliposis adenomatosa familiar originada por la nueva mutación W421X
Orestis Ioannidis1, George Paraskevas2, Stavros Chatzopoulos1, Anastasios Kotronis1, Nikolaos Papadimitriou1, Athina Konstantara1, Apostolos Makrantonakis1 and Emmanouil Kakoutis1
1First Surgical Department. General Regional Hospital George Papanikolaou. Thessaloniki, Greece.
2Department of Anatomy. Medical School. Aristotle University of Thessaloniki. Thessaloniki, Greece
ABSTRACT
Familial adenomatous polyposis (FAP) is a rare syndrome characterized by the presence of hundreds to thousands of colorectal adenomas and is responsible for less than 1% of all colorectal cancers. The syndrome is also characterized by extra-colorectal features including amongst others upper gastrointestinal tract polyps and desmoid tumors. The syndrome is inherited by an autosomal dominant gene, the adenomatous polyposis coli (APC) gene. We present the physical history, clinical presentation, diagnosis and treatment of a patient with a novel germline APC mutation, the W421X mutation, which resulted in FAP presenting with about a hundred colorectal polyps, gastric hyperplastic polyps and multiple aggressive intra-abdominal and extra-abdominal desmoid tumors.
Key words: Adenomatous polyposis coli. Colorectal cancer. Fibromatosis. Mutation.
Introduction
Familial adenomatous polyposis (FAP) while rare, as it is responsible for about 1-2% of all colorectal cancers, represents a distinctive syndrome both genetically and clinically (1-3). FAP is characterized by the presence of hundreds to thousands of colorectal adenomas which have an almost 100% possibility of malignant transformation by the 5th decade (1,2,4). The syndrome is also characterized by extra-colorectal features including upper gastrointestinal tract polyps, especially of the duodenum, desmoid tumors (fibromatosis), osteomas, epidermoid cysts, congenital hypertrophy of the retinal pigment epithelium, dental anomalies and increased risk for peri-ampullary carcinoma, meduloblastoma, papillary carcinoma of the thyroid and hepatoblastoma (1,2,5). The syndrome is inherited by an autosomal dominant gene, the adenomatous polyposis coli (APC) gene, although new mutations account for 15-30% of cases and results from a germline mutation (1,2,6,7). We present the physical history, clinical presentation, diagnosis and treatment of a patient with a novel germline APC mutation, the W421X mutation, which resulted in FAP presenting with about a hundred colorectal polyps, gastric hyperplastic polyps and multiple aggressive intra-abdominal and extra-abdominal desmoid tumors.
Case report
A 28 old year old female first presented with reported rectal bleeding for about 6 months. The digital rectal examination was positive for blood. The laboratory examination indicated anemia (HT: 31.5%, Hb: 9.9 g/dL). The barium enema demonstrated multiple polyps of the large bowel. The colonoscopy revealed the presence of multiple polyps of the large bowel from the rectum to the cecum, but especially of the sigmoid and transverse colon measuring form 2 mm to 3 cm (Fig. 1). Histopathological examination of the biopsies revealed tubular adenomas of the large bowel with moderate dysplasia. Gastroscopy demonstrated multiple hyperplastic polyps and the biopsy confirmed the hyperplastic nature of the gastric polyps (Fig. 2). Abdominal CT scan was with normal findings.


The family history revealed that the patient's mother has died from a brain metastasis from an unknown primary malignancy at the age of 52 and a maternal uncle deceased from generalized carcinomatosis from an inoperable colon cancer at the age of 56. The diagnosis of familial adenomatous polyposis was made and the patient was submitted to prophylactic total colectomy and ileorectal anastomosis with a J pouch. The patient was submitted to routine follow up with colonoscopy and gastroscopy and numerous stomach tubulovillous polyps have been removed.
The patient has been asymptomatic for 6 years but presented at that time with a mild colic abdominal pain for about a month. Laboratory examination was within the normal limit except of a mild anemia (Ht: 34,7%; Hb: 11 g/dL) and slightly elevated SCOT IU/L(41), SGPT IU/L (49) and amylase (124). Physical examination, gastroscopy and colonoscopy revealed the same findings as previous examinations. Preoperative electrocardiogram showed cardiac arrhythmia and the patient received treatment with mexitil.
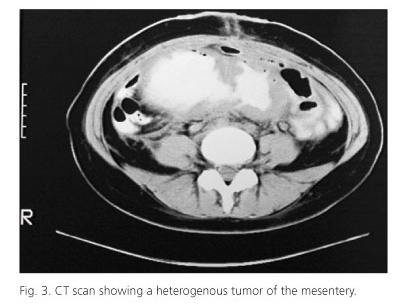
The abdominal ultrasound showed a large firm painless intraabdominal mass of the lower abdomen. The abdominal CT demonstrated a large heterogenous soft tissue intraabdominal mass measuring 16 x 8.5 x 13 cm that originated from the mesentery and seemed closely attached to the small bowel (Fig. 3). The mass at points displaced the small intestine but also entrapped and seemed to infiltrate the small intestine which proximal to the mass was dilated. In addition enlarged mesenteric and lumbar lymphnodes were noticed. Furthermore the right lobe of the liver had unhomogenous and subdense areas. The differential diagnosis included pseudomyxoma peritonei, mesenteric sarcoma, peritoneal carcinomatosis and abdominal fibromatosis. Percutaneous biopsies were not conclusive. An exploratory laparotomy was performed. The tumor was inoperable as it infiltrated the mesentery and excision wasn't fissile without excising almost whole the small intestine so only biopsies were taken (Fig. 4). The thirteenth postoperative day the patient presented fever, leukocytosis and abdominal pain. The abdominal CT showed free air and fluid subdiaphragmaticaly and right pleural effusion. The abscess was drained percutaneously under CT guidance with a pigtail catheter. Six months after the operation the patient presented with a left lower quadrant inflammation. The CT scan demonstrated an abscess which was successfully drained surgically.

After the diagnosis of intraabdominal desmoid tumor the patient received treatment with tamoxifen and non steroid anti-inflammatory drugs (sulindac 150 mg x 2) for 2 years. During that period the desmoid tumor remained stable in size. However 2 years after the initial diagnosis another mass developed at the level of the uterus and the urinary bladder while the mesenteric desmoid tumor has subsided to desmoplastic reaction of the mesentery (Fig. 5). Treatment was changed to rofecoxib 25 mg x 1 with tamoxifen for about a year but 3 years after the diagnosis of desmoids tumor another tumor started developing in the left rectus abdominis muscle (Fig. 6). Treatment was once again changed to valdecoxib 200 mg 1 x 1 and tamoxifen but the tumors continued growing. Four years after the diagnosis of desmoid tumor their started treatment with imatinib (400 mg x 2) for 8 months. The patient presented an initial improvement but soon after presented resistance to treatment despite increased dosage and the tumors continued growing. Following the patient received treatment with sunitinib 50 mg per day. And presented clinical and symptomatic improvement.


Eleven years after diagnosis of FAP and 5 after the development of desmoid tumors the patient was submitted to genetic testing for APC mutation. A W421X mutation was found. This mutation causes the codon of tryptophan to change to a stop codon thus resulting in early ending of protein synthesis and deactivation of one allele.
The patient died 11.5 years after the initial diagnosis due to cachexia, gastrointestinal bleeding and cardiac arrest.
Discussion
Desmoid tumors are rare benign soft tissue tumors that don't metastasize (1,7,8) but tend to invade locally and compress surrounding structures (1,7,8). Desmoid tumors may appear sporadically but present a known association with familial adenomatous polyposis (8,9). Specifically, in general population they are rare and account for only 0.03% of all neoplasms while their incidence in FAP varies greatly from 3.5 to 32%.
The exact etiology of desmoid tumors is poorly defined although several factors are acknowledged to be positively correlated with their development and growth (7,9). Trauma, and especially surgical, seems to be a potential initiating factor in desmoids tumor development (7,8,9). In addition, estrogens have been considered to be involved in the pathogenesis of desmoid tumors. Also, the close association of desmoid tumor and FAP reveals a genetic predisposition and the position of the APC germline mutation is considered to be an important factor affecting the likelihood of developing desmoids (7,8,9). Mutation in codon 1444 or beyond seems to be associated with increased risk of desmoids growth (7,8). In the current patient despite the fact that the mutation was in codon 421 the patient developed multiple aggressive desmoid tumors resistant to treatment.
Desmoid tumors are fibroblastic mesenchymal cell monoclonal proliferations that can arise from musculoaponeurotic and fascial structures (8,9). Macroscopically desmoids are hard fibrous homogenous lumps, with pale tan color usually grayish-white. They are well defined but lack a true capsule and frequently infiltrate surrounding tissues. Microscopically the tumors are consisted of a dense collagenous matrix with highly differentiated fibroblasts and bundles of myofibroblasts. The amount of mitoses is normal and nuclei are regular and small (7-9).
The anatomical location of desmoid tumors in patients with FAP is the abdominal cavity in about half the patients, the abdominal wall in 40% and in about 10% the extremities (7). Most of the intraabdominal desmoids (85-100%) are located in the mesentery and the rest (15%) in the retroperitoneum (7). Multiple desmoid tumors both intraabdominal and in the abdominal wall are rare (10). Distribution is different for sporadic cases of desmoid tumors (7,8). Clinically desmoids may present with a wide range of symptoms from asymptomatic slowly growing small tumors to large rapidly-growing masses causing symptoms related to small bowel obstruction and ureteric compression (7,8) and leading to death in a matter of a few years or even months (8). Regarding clinical course the tumors can resolve spontaneously, remain stable, and undergo cycles of progression and resolution or progress rapidly (8). In the present case the patient initially presented with a mesenteric desmoid tumor causing partial small bowel obstruction but subsequently developed multiple tumors one tumor between the uterus and urinary bladder and another in the abdominal wall while the tumor of the mesentery has resolved.
The diagnosis of desmoid tumors is often clinical but imaging studies are helpful in establishing the degree of local infiltration and receiving percutaneous biopsies which will confirm the diagnosis. Regarding abdominal and intraabdominal desmoids CT and MRI have proven useful in order evaluate extension, predict resectability and monitor the treatment outcome (7-9).
Treatment of the desmoid tumors is mainly empirical. Simple observation of asymptomatic tumors is one viable option (7,8). Surgery is considered the treatment of choice for extremity and abdominal wall desmoid tumors but for intraabdominal desmoids radical excision is often not feasible and even in cases of resection with uninvolved margins the recurrence rate is high ranging from 10-68%. Also surgery is associated with high percentages of complications and increased post-operative mortality and morbidity (7-9). Medical treatment may prove useful especially in patients with FAP and desmoid tumors. Anti-estrogens and non-steroid anti-inflammatory drugs (NSAIDS) represent the mainstay of non-cytotoxic chemotherapy. Amongst the NSAIDS sulindac in a daily dosage of 200 to 400 mg has been used widely with variable results. Cyclooxygenase 2 inhibitors, including celecoxib, may prove useful. However, in the present case none of the cyclooxygenase 2 inhibitors administrated -valdecoxib and rofecoxib, which have both been withdrawn- was effective in the treatment of the tumors. NSAIDS are administrated usually in combination with anti-estrogens, mostly tamoxifen 20-40 mg daily or toremifen 180 mg daily, based on the observation that desmoid tumors demonstrate estrogen receptors (7-9). Cytotoxic chemotherapy is related with considerable morbidity and mortality and should only be reserved for patients not responding to NSAIDS and anti-estrogen (7-9). Radiotherapy has no role in intraabdominal desmoid tumors (7-9). Recently, imatinib, a kinase inhibitor, has been used in two clinical trials for the treatment of desmoid tumors and may have a role in the management of unresectable tumors leading to long term stable disease in a large portion of patients (11,12). In patients resistant to imatinib, sunitinib may be an effective alternative as the spectrum of tyrosine kinase inhibited is broader than that of imatinib (13). In the present case imatinib seemed to have stopped progression of the tumor of about 6 months but then was rendered ineffective. Sunitinib also seemed to be ineffective as the tumors progressed and the patient died from progressive disease under sunitinib treatment.
In conclusion, desmoid tumors are benign but highly invasive tumors which appear with increased frequency in patients with FAP. Their etiology as well as the treatment of choice is not yet fully understood. Our case is the first case of a FAP patient bearing the W421X mutation in whom multiple aggressive desmoid tumors developed which were resistant to all medical treatments, including NSAIDS, anti-estrogens and tyrosine kinase inhibitors.
References
1. Lipton L, Tomlinson I. The genetics of FAP and FAP-like syndromes. Fam Cancer 2006;5:221-6. [ Links ]
2. Jass JR. Colorectal polyposes: from phenotype to diagnosis. Pathol Res Pract 2008;204:431-47. [ Links ]
3. Fernández-Suárez A, Cordero Fernández C, García Lozano R, Pizarro A, Garzón M, Núñez Roldán A. Clinical and ethical implications of genetic counselling in familial adenomatous polyposis. Rev Esp Enferm Dig 2005;97:654-65. [ Links ]
4. Garzón-Benavides M, Pizarro-Moreno A, García-Lozano R, Herrero-Garrido MI, Hervás-Molina AJ, Márquez-Galán JL, et al. Andalusian Registry for familial adenomatous polyposis. Analysis of patients included. Rev Esp Enferm Dig 2010;102:653-7. [ Links ]
5. Perea J, Justo I, Alvaro E, Lomas M, Tasende JD, Marín JC, et al. Surgical management of hereditary colorectal cancer: surgery based on molecular analysis and family history. Rev Esp Enferm Dig 2009; 101:536-40. [ Links ]
6. Herráiz M, Muñoz-Navas M. Recognition and management of hereditary colorectal cancer syndromes. Rev Esp Enferm Dig 2009;101:125-32. [ Links ]
7. Knudsen AL, Bülow S. Desmoid tumour in familial adenomatous polyposis. A review of literature. Fam Cancer 2001;1:111-9. [ Links ]
8. Sturt NJ, Clark SK. Current ideas in desmoid tumours. Fam Cancer 2006;5:275-88. [ Links ]
9. Shields CJ, Winter DC, Kirwan WO, Redmond HP. Desmoid tumours. Eur J Surg Oncol 2001;27:701-6. [ Links ]
10. Heiskanen I, Järvinen HJ. Occurrence of desmoid tumours in familial adenomatous polyposis and results of treatment. Int J Colorectal Dis 1996;11:157-62. [ Links ]
11. Chugh R, Wathen JK, Patel SR, Maki RG, Meyers PA, Schuetze SM, et al. Sarcoma Alliance for Research through Collaboration (SARC). Efficacy of imatinib in aggressive fibromatosis: results of a phase II multicenter Sarcoma Alliance for Research through Collaboration (SARC) trial. Clin Cancer Res 2010;16:4884-91. [ Links ]
12. Penel N, Le Cesne A, Bui BN, Perol D, Brain EG, Ray-Coquard I, et al. Imatinib for progressive and recurrent aggressive fibromatosis (desmoid tumors): an FNCLCC/French Sarcoma Group phase II trial with a long-term follow-up. Ann Oncol 2011;22:452-7. [ Links ]
13. Skubitz KM, Manivel JC, Clohisy DR, Frolich JW. Response of imatinib-resistant extra-abdominal aggressive fibromatosis to sunitinib: case report and review of the literature on response to tyrosine kinase inhibitors. Cancer Chemother Pharmacol 2009;64:635-40. [ Links ]
![]() Correspondence:
Correspondence:
Orestis Ioannidis.
Thessaloniki, Greece.
e-mail: telonakos@hotmail.com
Received: 04-08-11.
Accepted: 13-09-11.

