










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.109 no.6 Madrid jun. 2017
https://dx.doi.org/10.17235/reed.2017.4167/2015
CASE REPORT
The clinical extremes of autoimmune cholangitis
Sara Campos1, Dário Gomes1, Maria Augusta Cipriano2 and Carlos Sofia1
Departments of 1Gastroenterology and 2Pathology. Centro Hospitalar e Universitário de Coimbra. Coimbra, Portugal
ABSTRACT
Autoimmune cholangitis (AIC) was first described in 1987 as immunocholangitis in three women who presented with signs and symptoms of primary biliary cholangitis (PBC), but who were antimitochondrial (AMA) negative and antinuclear antibodies (ANA) positive, and responded to immunosuppressive therapy with azathioprine and prednisolone (1). AIC is a rare chronic cholestatic inflammatory disease characterized by the presence of high ANA or smooth muscle antibodies (SMA) but AMA seronegativity. Histologically, AIC exhibits bile duct injury (2). In terms of therapeutics, in addition to response to ursodeoxycholic acid, a prompt response to corticosteroids has also been reported in earlier stages, distinguishing it from PBC.
Herein the authors describe two cases with mixed signs of PBC and autoimmune hepatitis (AIH). The diagnostic differentiation between these diseases (AIC, PBC and AIH) is essential because of the different therapeutic strategies. Our cases highlight the importance of clinician awareness of the autoimmune spectrum of liver diseases.
Key words: Autoimmune cholangitis. Autoimmune hepatitis. Primary biliary cholangitis. Cholestatic liver disease.
Case report
Case report 1
A 56-year-old female patient was referred to the Department of Gastroenterology of Centro Hospitalar e Universitário de Coimbra for asymptomatic cholestasis detected during a peri-menopausal stage. No personal or family history of liver disease was present. She denied regular medications, alcohol or drug abuse, blood transfusions or previous surgeries. Physical examination was unremarkable.
Laboratory tests revealed alkaline phosphatase (AP) at 155 (30-120) U/l, gama-glutamyltranspherase (GGT) at 339 (< 55) U/l, aspartate aminotranspherases (AST) at 129 (< 35) U/l, alanine aminotranspherases (ALT) at 169 (< 45) U/l and total cholesterol at 211 (< 200) mg dl. Other laboratory markers including serum bilirubin, albumin, prothrombin time, ferritin, transferrin saturation and ceruloplasmin were normal. Serum HBsAg, anti-HBc, anti-HBs and HCV antibodies were negative.
Abdominal ultrasound (Fig. 1) did not show any changes, namely signs with regard to chronic liver disease, focal lesions or bile duct obstruction.

Laboratory tests also showed ANA at 1:320, AMA and SMA antibodies negative, gamma-globulin at 1.07 (0.7-1.5) g/l, immunoglobulin-M at 3.53 (0.46-3.04) g/l and immunoglobulin-G at 9.95 (7.51-15.6) g/l.
Liver biopsy (Fig. 2) showed granulomatous cholangitis, focal bile duct loss and signs of chronic cholestasis in periportal hepatocytes (rhodanine stain) suggesting the diagnosis of autoimmune cholangitis.
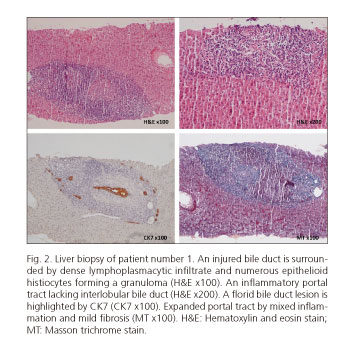
Combination therapy with a per os prednisolone cycle followed by ursodeoxycholic acid (UDCA) 750 mg per day was prescribed, with resolution of the biochemical abnormalities. She is currently on UDCA at 750 mg per day.
Case report 2
A 41-year-old female patient was referred to the Department of Gastroenterology of Centro Hospitalar e Universitário de Coimbra for cholestasis, accompanied by chronic, generalized and intense pruritus, which was worse at night, affecting sleep and fatigue. No personal or family history of liver disease was reported. She denied regular medications, alcohol or drug abuse, blood transfusions or surgeries. Physical examination was normal.
Laboratory tests revealed AP at 523 (30-120) U/l, GGT at 299 (< 55) U/l, AST at 117 (< 35) U/l, ALT at 196 (< 45) U/l and total cholesterol at 268 (< 200) mg/dl. Other laboratory markers including serum bilirubin, albumin, prothrombin time, ferritin, transferrin saturation and ceruloplasmin were normal. Markers of hepatitis B and C were negative.
Abdominal ultrasound and MRCP (Fig. 3) documented no focal lesions or dilatation of biliary tract.

ANA was at 1:160 and AMA and SMA antibodies were negative; gamma-globulin was at 0.81 (0.7-1.5) g/l; immunoglobulin-M, at 3.7 (0.46-3.04) g/l; and immunoglobulin-G, at 11.9 (7.51-15.6) g/l.
Liver biopsy (Fig. 4) described "incomplete septal fibrosis and a chronic hepatitis with moderate activity, ductopenia and histological signs of chronic cholestasis, pointing to a chronic cholestatic syndrome type autoimmune cholangitis".

Multiple therapies were administered in this patient (UDCA up to 1,000 mg per day, cholestyramine up to 16 mg per day, rifampicin up to 600 mg per day, naltrexone up to 50 mg per day, sertraline up to 75 mg per day, hydroxyzine up to 25 mg four times a day, amitriptyline up to 25 mg per day, phototherapy, molecular adsorbent recirculating system sessions, prednisolone up to 30 mg per day and budesonide up to 6 mg per day, azathioprine up to 75 mg per day, mycophenolate mofetil up to 1,500 mg per day) without significant laboratory or clinical improvement. Her last transient elastography showed fibrosis grade 4. She is currently waiting for a liver transplant.
Discussion
As demonstrated with these cases, AIC can be difficult to diagnose because of its biochemical, serological and histological similarities with other entities. All epidemiological (sex and age), clinical, biochemical and histological findings, as well as response to UDCA (as in the first patient), could suggest PBC. Nevertheless, the absence of detectable AMA in combination with high titers of ANA could also indicate type-1 autoimmune hepatitis (AIH) with a predominantly cholestatic course. Against this hypothesis, however, is the fact that elevation of serum GGT and/or AP were disproportionate in comparison to ALT/AST elevation, indicating lower grade hepatocellular injury, and gamma-globulin IgG was not increased. Moreover, in the first case, the maintenance of remission was achieved when combining corticosteroids with UDCA, and in the second case, the immunosuppressive therapy was insufficient to decrease the levels of transaminases.
AIC is an enigmatic entity, with the knowledge of its etiopathogenesis still evolving. Some authors defend it as a separate entity with varying manifestations (2-7), while others agree on the fact that it is a variant of PBC (AMA-negative PBC) (2-5). Concordant to this second theory is the fact that these entities share a female preponderance, symptoms such as fatigue and pruritus, a cholestatic serum enzyme pattern, florid bile duct lesions and a slowly progressive course leading to fibrosis and liver cirrhosis (6). All in all, 5-10% of PBC patients can also be ANA or SMA positive and AMA negative (6). More recently, the presence of pyruvate dehydrogenase complex on the biliary epithelia (PDC subunit E2), considered as an immunodominant autoantigen in PBC, potentiates this theory. Moreover, the presence of antibodies directed to 2 oxo-acid-dehydrogenase complex (2-OADC) is in favor of the same origin. However, in contrast to PBC, CAI may have different HLA, lower immunoglobulin-M and AST levels. The presence of anti-lactoferrin and antibodies against carbonic anhydrase, an enzyme found in epithelial cells including the bile duct which is known to promote choleresis, previously supporting the idea of CAI being an independent entity is now known to be non-specific and even found in healthy individuals. Some more remote opinions consider AIC as a cholangiopathic variant of AIH (2-6) or a result of a transitional stage of these two classical diseases (AIH and PBC) (2,5). AIC prevalence is unknown, but it is estimated that 11% of the adults with PBC or AIH may belong to this category (7).
A distinction must be made between AIC and overlap syndromes between PBC and AIH. This latter syndrome, first reported in the 1970s (7) and already reported in almost 10% of adults with AIH and PBC (8), typically shows features of both diseases, with a significant increase in transaminases and cholestasis parameters, high titers of AMA in combination with high titers of ANA and AML, and histological findings resembling both diseases (7,9). The biochemical and serological hallmarks for this classical overlap syndrome were absent in these cases, as well as the histopathological features. Therefore, we can conclude that our patients seem to belong to the AIC group.
One patient had no initial complaints, in contrast to the second. Ben-Ari has already shown that AIC can present with a wide clinical spectrum between asymptomatic and liver failure (3).
The lack of response to immunosuppressive therapy and treatment with UDCA observed in the second case has also been already reported in other small case series (10). Unfortunately, there is still no consensus regarding AIC therapy because controlled prospective clinical trials have not been conducted.
Our cases thus highlight the importance of the clinician to be aware of the autoimmune spectrum of liver diseases (AIC, PBC, AIH, overlap syndrome PBC-AIH), so that an appropriate therapeutic strategy can be adopted in each case.
References
1. Brunner G, Klinge O. A cholangitis with antinuclear antibodies (immunocholangitis) resembling chronic destructive non-suppurative cholangitis. Dtsch Med Wochenschr 1987;112:1454-8. [ Links ]
2. Czaja AJ. The variant forms of autoimmune hepatitis. Ann Intern Med 1996;125:488-598. DOI: 10.7326/0003-4819-125-7-199610010-00009. [ Links ]
3. Ben-Ari Z, Dhillon AP, Sherlock S. Autoimmune cholangiopathy: Part of the spectrum of autoimmune chronic active hepatitis. Hepatology 1993;18:10-5. DOI: 10.1002/hep.1840180103. [ Links ]
4. Omagari K. Autoimmune cholangitis: Is it an antimitochondrial antibody negative primary biliary cirrhosis? Acta Med Nagasaki 1999;44(1-2):11-4. [ Links ]
5. Czaja AJ, Carpenter HA, Santrach PJ, et al. Autoimmune cholangitis within the spectrum of autoimmune liver disease. Hepatology 2000;31:1231-8. DOI: 10.1053/jhep.2000.7878. [ Links ]
6. Sharma B, Raina S, Sharma R. Autoimmune cholangitis: A variant syndrome of autoimmune hepatitis. Case Reports Hepatol 2014;2014:501530. DOI: 10.1155/2014/501530. [ Links ]
7. Beuers U, Rust C. Overlap syndromes. Semin Liver Dis 2005;25(3):311-20. DOI: 10.1055/s-2005-916322 [ Links ]
8. Sabite K, Erkan P, Yasemin K, et al. Autoimmune cholangitis: Six cases. Turk J Gastro 2000;(11):4. [ Links ]
9. EASL Clinical Practice Guidelines: Management of cholestatic liver diseases. J Hepatol 2009;51:237-67. [ Links ]
10. Mohr L, Heintges T, Hensel F, et al. Treatment of autoimmune cholangitis. Dig Dis Sciences 1999;(43)9:2160-3. [ Links ]
![]() Correspondence:
Correspondence:
Sara Teles de Campos.
Department of Gastroenterology.
Centro Hospitalar e Universitário de Coimbra.
Praceta Prof. Mota Pinto.
3000-075 Coimbra, Portugal
e-mail: saratcampos@gmail.com
Received: 19-12-2015
Accepted: 17-12-2016
