










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista Española de Enfermedades Digestivas
versión impresa ISSN 1130-0108
Rev. esp. enferm. dig. vol.109 no.11 Madrid nov. 2017
https://dx.doi.org/10.17235/reed.2017.5044/2017
Cystic pancreatic neuroendocrine tumors (cPNETs): a systematic review and meta-analysis of case series
Tumores quísticos neuroendocrinos de páncreas (cPNET): revisión sistemática y metaanálisis de series de casos
Luis Hurtado-Pardo1, Javier A. Cienfuegos1-3, Miguel Ruiz-Canela2,3, Pablo Panadero4, Alberto Benito5 and José Luis Hernández-Lizoain1
1Department of General Surgery. Clínica Universidad de Navarra. Pamplona, Spain.
2Department of Preventive Medicine and Public Health. Universidad de Navarra. Pamplona, Spain.
3Institute of Health Research of Navarra (IdisNA). Pamplona, Spain.
4Department of Pathology, and 5Department of Radiology. Clínica Universidad de Navarra. Pamplona, Spain
ABSTRACT
Cystic pancreatic neuroendocrine tumors represent 13% of all neuroendocrine tumors.
The aim of this study is to analyze the phenotype and biologic behavior of resected cystic neuroendocrine tumors.
A systematic review and meta-analysis were conducted until September 2016 using a search in Medline, Scopus, and EMBASE with the terms "cystic pancreatic endocrine neoplasm", "cystic islets tumors" and "cystic islets neoplasms".
From the 795 citations recovered 80 studies reporting on 431 patients were selected. 87.1% (n = 387) were sporadic tumors and 10.3% (n = 40) corresponded to multiple endocrine neoplasia type 1. Were diagnosed incidentally 44.6% (n = 135). Cytology was found to have a sensitivity of 78.5%.
Were non-functional tumors 85% (n = 338), and among the functional tumors, insulinoma was the most frequent. According to the European Neuroendocrine Tumor Society staging, 87.8% were limited to the pancreas (I-IIb), and 12.2% were advanced (III-IV).
Disease-free survival at 5 years in stages (I-IIIa) and (IIIb-IV) was 91.5% and 54.2%, respectively; and was significantly lower (p = 0.0001) in functional tumors. In patients with multiple endocrine neoplasia there was a higher incidence of functional (62.5%) and multifocal (28.1%) tumors. Disease-free survival at 5 and 10 years was 60%.
Cystic pancreatic neuroendocrine tumors exhibit phenotypical characteristics which are different to those of solid neuroendocrine tumors.
Key words: Cystic pancreatic neuroendocrine tumors. Multiple endocrine neoplasia type 1. Cystic tumors. Oncologic outcomes. Evidence base medicine.
RESUMEN
Los tumores quísticos neuroendocrinos representan entre el 13% de los tumores neuroendocrinos de páncreas.
El objetivo del trabajo es realizar una revisión sistemática y un metaanálisis de las series de casos descritas.
Se realizó una revisión sistemática hasta septiembre de 2016 mediante una búsqueda en Medline, Scopus y EMBASE con los términos: "cystic pancreatic endocrine neoplasm", "cystic islets tumors" y "cystic islets neoplasms".
De 795 citas se seleccionaron 80 estudios que describían 431 pacientes, incluyendo 5 casos propios. El 87,1% (n = 387) eran tumores esporádicos y el 10,3% (n = 40) correspondían a neoplasia endocrina múltiple tipo 1. El 44,6% (n = 135) fueron diagnosticados de forma incidental. La citología mostró una sensibilidad del 78,5%.
El 85% (n = 338) eran tumores no funcionantes; y el insulinoma fue el más frecuente entre los funcionantes. Según la estadificación European Neuroendocrine Tumor Society, el 87,8% estaban limitados al páncreas (I-IIb) y el 12,2% eran avanzados (III-IV).
La supervivencia libre de enfermedad a los 5 años en estadios (I-IIIa) y en los estadios (IIIb-IV) fue del 91,5% y 54,2% respectivamente; y fue significativamente menor (p = 0,0001) en los tumores funcionantes. En los pacientes con MEN-1 hubo mayor incidencia de funcionantes (62,5%) y multicéntricos (28,1%).
Los tumores quísticos neuroendocrinos de páncreas expresan un fenotipo diferente a los tumores endocrinos sólidos, pero tienen un pronóstico similar, tras la resección a excepción de los tumores hereditarios.
Palabras clave: Tumores quísticos neuroendocrinos páncreas. Neoplasia endocrina múltiple tipo 1. Tumores quísticos. Resultados oncológicos. Medicina basada evidencia.
Introduction
Cystic pancreatic neuroendocrine tumors (cPNETs) account for between 13% and 17% of pancreatic neuroendocrine tumors (PNETs) and 9-10% of resected cystic tumors (1,2).
Due to the widespread use of diagnostic methods with greater sensitivity and specificity the incidence of such tumors has risen in recent years and has led to the publication of cases and clinical series.
Establishing a differential diagnosis with regard to other cystic and solid lesions of the pancreas may be difficult as in many cases cPNETs are diagnosed incidentally and exhibit great clinical variability: functional, non-functional, sporadic or hereditary (3-5).
The aim of the present study is to carry out a systematic review and meta-analysis of resected cystic pancreatic neuroendocrine tumors reported in the literature, including 5 cases from our own center, with the objective of obtaining the most exhaustive scientific knowledge on their clinical, pathologic and prognostic characteristics.
Material and methods
The systematic review was carried out following the recommendations reported in the "Preferred Reporting Items for Systematic Reviews and Meta-Analyses" (PRISMA) adapted to surgical series and cases (6,7).
Cystic pancreatic neuroendocrine tumor was defined as any neuroendocrine tumor which had a cystic pattern (purely cystic or mixed cystic-solid lesions) in the macroscopic study or the imaging studies and confirmed by histology.
- Inclusion criteria: all published cases of cystic pancreatic neuroendocrine tumors which were treated with surgery and with histologic confirmation were identified. Non-resected tumors or those where histologic confirmation was lacking were excluded.
- Methods use to search for and identify studies: up to 22nd September 2016, the following databases were consulted: Medline, Scopus, and EMBASE. Studies in English, German French and Spanish were included. A search of the "grey" literature was also undertaken with the inclusion of cases reported at conferences, letters to the editor or chapters in books. The search terms used were "cystic pancreatic endocrine neoplasms", "cystic islets tumors", and "cystic islets neoplasms".
The articles identified were recorded in an Excel® database and duplicates were eliminated. In the database, the titles and abstracts of the articles were independently reviewed by two authors (LH, JAC) in order to assess the validity and quality of the case series. The full text of all the articles selected was printed to ensure the inclusion criteria had been met. When doubts arose, the possible reasons for excluding the article were discussed by the team, and a final decision was taken by consensus. If multiple case series were published from the same center with overlapping study periods, the publication with the largest number of cases was selected.
-Data extraction: data were extracted using a predefined protocol which included sex, age, associated genetic disorder, symptoms, presentation type, diagnostic methods, fine needle punction-aspiration (FNPA) cytology, size and location of the tumor, type of surgery, post-operative complications, immunohistochemical and pathologic characteristics, stage according to ENETS (European Neuroendocrine Tumor Society) (8), Ki-67 grade as defined by the WHO (World Health Organization) (9), survival and time to tumor recurrence. All data were extracted in duplicate to ensure accuracy.
- Data synthesis and analysis: The continuous variables were summarized with means, ranges and standard deviations. In the series of patients where individual patient data were not provided, these data were not included in the analysis. Categorical variables were analyzed using Pearson's χ2 test or Fisher's exact test. To compare continuous variables from independent samples the Student's t-test was used or its nonparametric equivalent, the Mann-Whitney U test. The two-sided level of significance was set at p < 0.05.
Kaplan-Meier curves were plotted to assess survival, with a significance level of p < 0.05, and using the Log-rank test to analyze differences in survival depending on different characteristics.
All data were analyzed using Stata 12 software (StataCorp. Stata Statistical Software: Version 12.1. College Station, TX: Stata Corp LP).
Results
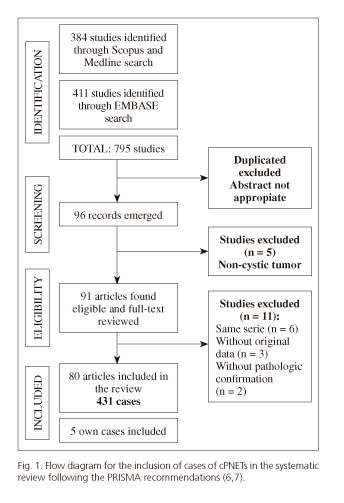
A total of 795 citations were identified. Once duplicate and unrelated articles had been excluded, 96 studies were selected, from which another 5 were discarded as they did not deal with cystic PNETs. Of the remaining 91 articles, a further 11 were excluded: 6 articles because they reported on the same series included in another article, 3 cases because they provided no data and 2 cases in which pathologic confirmation of the tumor was not performed. Finally, 80 studies were included, 67 isolated case reports (10-76) and 13 case reviews (1,2,77-87). Of these 80 studies, the full text was not available in 15, and so the relevant information was taken from the abstract. The 80 studies covered a total of 431 cases of cystic pancreatic neuroendocrine tumors. Five cases who underwent surgery in our center were also included to make a final total on 436 cPNET cases. Figure 1 shows the flow diagram of the articles included and reasons for exclusion as set out in the PRISMA guidelines (6).
The demographic and clinical characteristics of the 436 patients are shown in table 1. Of these patients 194 (50.9%) were male, and the mean age was 53.2 years (SD = 16.1). Male patients were significantly older (57.1 vs. 49.5; p = 0.001).


Genetic characteristics were reported in 387 patients: 337 patients (87.1%) were sporadic, 40 (10.3%) had MEN-1 (multiple endocrine neoplasis type 1), 7 (1.2%) had Von Hippel Lindau syndrome and 3 tuberous sclerosis (0.8%).
Details of the clinical presentation were obtained in 303 patients, of whom 135 (44.6%) were asymptomatic and thus corresponded to "incidentalomas", 106 (35%) presented with abdominal pain, 16 (15.3%) with an abdominal mass, 14 (4.6%) with weight loss, 10 (3.3%) diabetes on diagnosis and 8 (2.6%) pancreatitis.
Data on the functionality of the tumors were available in 339 cases. In 287 cases (85%) they were non-functional while in 52 (15.3%) they were functional: 18 insulinomas (5.3%), 12 glucagonomas (3.5%), 10 gastrinomas (3%), 7 somatostitomas (2.1%), 3 ACTH-secreting tumors (ACTHoma) (0.9%), 1 vasoactive intestinal peptide-producing tumor (VIPoma) (0.3%) and 1 growth hormone-producing tumor (GHoma) (0.3%).
In 196 patients a preoperative diagnosis was made using FNPA leading to a diagnosis of endocrine tumor in 154 patients (78.6%) and inconclusive results in 20 cases (10.2%).
Tumor location was reported in 374 patients. Approximately half of the tumors, 184 (49.2%) were located in the tail, 87 (23.3%) in the head, 77 (20.6%) in the body, 7 (1.9%) in the neck and 6 (1.6%) in the uncinate process. Thirteen cases (3.5%) were multifocal tumors. The maximum diameter reported in 193 patients was 43.8 mm (SD = 36.5).
The type of surgical intervention was reported in 221 patients: in 28 cases (12.7%) the procedure performed was enucleation, in 31 cases (14%) Whipple's procedure, in 20 cases (9.1%) corporocaudal pancreatectomy, in 7 cases (3.2%) central pancreatectomy, in 127 cases (57.5%) distal pancreatectomy, in 7 cases (3.2%) total pancreatectomy and in 1 case marsupialization. In 18 cases the procedures were performed laparoscopically.
Information on ENETS staging was available for 269 patients: 69 (25.7%) were stage I, 98 (36.4%) stage IIa, 69 (25.7%) stage IIb, 2 (0.7%) stage IIIa, 18 (6.7%) stage IIIb and 13 (4.8%) stage IV. Lymph node involvement was reported in 295 patients with 268 cases (90.8%) with N0 and the remaining 27 (9.2%) with N1. Synchronous metastases were found in 13 patients (4.6%) of the 286 for whom this information was given.
The mitotic index as defined by Ki-67 was reported in 218 of the tumors and was low in 186 (85.3%) and intermediate in 32 (14.9%). Twenty-two foci of necrosis (n = 163) and 55 foci of hemorrhage (n = 185) were identified.
Follow-up was reported in 187 cases with a mean duration of 42.5 months (SD = 30.3) and a range of 1 to 168 months. Out of these 187 cases, there were 15 cases of relapse (8%): 5 (2.8%) were locoregional, 4 (2.1%) distant and in 6 cases (3.2%) no location was given. Overall survival (OS) could be obtained for 185 cases, and 4 deaths were recorded (2.2%).
Overall survival as calculated by ENETS staging in stages I and II at 5 and 10 years was 100%. In stage III, OS at 1 and 5 years was 100% and 85.7%, respectively. In stage IV, OS at 1 and 5 years was 100% and 75% respectively (p = 0.05). Disease-free survival rates (DFS) at 1 and 5 years in stages I-IIIa (without lymph node involvement) were 98.5%, 91.5% and 91.5% respectively while in stages IIIb-IV (with lymph node involvement) DFS at 1, 5 and 6 years was 100%, 54.2% y 54.2% respectively (p = 0.001) (Fig. 2).

Analyses by specific subgroup
Functional vs. non-functional tumors
Table 2 summarizes the demographic and clinical characteristics of the 339 cases for whom the degree of functionality was reported. We found no significant differences regarding mean age at presentation between the two tumor types (49.2 vs. 50.3) (p = 0.896).

Of the 35 functional tumors, 12 (34.3%) corresponded to patients with MEN-1, while in the group of 262 patients with non-functional tumors 20 were identified as having MEN-1 (7.7%) (p = 0.001).
When location was compared, a greater proportion of multifocal tumors was found in the functional tumor group as compared to the non-functional group: 6 multifocal cases (17.6%) in the functional group (n = 34) as opposed to 7 multifocal cases (2.7%) in the non-functional groups (n = 260) (p = 0.016). No significant differences were found regarding size: in the non-functional tumor group (n = 84) mean tumor size was 51mm (SD = 46.3) in contrast with 45.8mm (SD = 31.6) in the functional tumor group (n = 27) (p = 0.984).
Comparison of both groups with regard to ENETS staging revealed that 41 cases (26.3%) were in stage I, 95 cases (60.9%) in stage II, 14 cases (9%) in stage III and 6 cases (3,8%) in stage IV in the non-functional group, whereas in the functional group there were 5 cases (15%) in stage I, 17 cases (51%) in stage II, 4 cases (12%) in stage III and 7 cases (21%) in stage IV (p = 0.007).
Overall survival was similar in both groups (p = 0.68). In contrast, when we compared DFS in the non-functional and functional cPNET, we found that patients with non-functional tumors had a significantly greater survival (p = 0.0001) than those with functional tumors.
MEN-1 patients
Table 3 summarizes the characteristics of the 40 cases of hereditary MEN-1. The mean age of the 27 patients with data was 41.7 years (SD=15). As for the degree of functionality, 20 patients (62.5%) had non-functional tumors and 12 (38.7%) functional tumors: 5 insulinomas (15.6%), 5 gastrinomas (15.6%), 1 glucagonoma (3.1%) and 1 GHoma (3,1%). Five cases (15.6%) were located in the head, 6 (18.8%) in the body, 12 (37.5%) in the tail and 9 (28.1%) were multifocal. The mean diameter reported for 26 of the patients was 40.2 mm (SD = 29.9).

According to the ENETS classification (n=26), 7 cases (26.9%) were stage I, 16 (61.5%) stage II, 2 (7.7%) stage III and 1 (3.8) stage IV. The Ki-67 index score was low in the 18 cases for whom this information was available.
Follow-up was recorded for 25 cases, with a mean of 46.8 months (SD = 39.6) and a range of 1 to 120 and 7 relapses (28%) and 1 death (4%) occurred.
Overall survival at 1, 5 and 10 years in this group of patients with MEN-1 was 100%, 91.7% and 91.7%, respectively. DFS at 1, 5 and 10 years was 100%, 60% and 60%, respectively.
Discussion
cPNETs account for 13-17%% of pancreatic neuroendocrine tumors and 9-10% of resected cystic tumors (1,2). Given their low incidence, their biological behavior is largely unknown. Although the first reports date from 1940, such tumors were not recognized as a specific entity until 2008 (1,88). Most cases have been reported as isolated cases, and only 13 authors have published series with more than 10 cases (1,2, 77-87).
In the literature two well defined periods can be identified: up until 2000 reports were of large symptomatic tumors and from 2000 onwards there was a predominance of small tumors, diagnosed incidentally due to the use of more sensitive imaging techniques, as a result of which there has been an increase in pancreatic "incidentalomas" and their management has generated certain controversy (89).
On the occasion of treating 5 cases in our center, and with the aim of investigating the phenotype and oncologic outcomes of these tumors, we performed a systematic review and a meta-analysis of published cases.
The etiopathogenesis of cPNETs is controversial. Kamisawa et al. (62) proposed that the slow, expansive growth of neuroendocrine tumors led to the development of a fibrous capsule compromising blood flow to the tumors and giving rise to infarction and central necrosis. Similarly, Buetow et al. (87) in a series of 133 pancreatic neuroendocrine tumors concluded that the presence of cystic degeneration or necrosis was correlated with the size of the tumor. In contrast, Iacono et al. (53) suggested that hemorrhage is the initial event in the development of the tumor's cystic form. Other authors propose that cPNETs are caused by the intraductal growth of a neuroendocrine tumor.
Furthermore, pathologic staging of pancreatic neuroendocrine tumors was not published until 2010 (ENETS) (8), which also led to confusion until the classification was widely adopted.
X-ray diagnosis has low specificity as the images are similar to those of other cystic lesions of the pancreas such as solid pseudopapillary tumors, mucinous tumors, intraductal mucinous papillary neoplasms and pancreatic metastases, among others. In the review by Singhi et al. (1) 43% of cPNETs were wrongly diagnosed. Khashab et al. (90) reported the diagnostic superiority of ultrasound endoscopy with an increase in yield over computed tomography of 36% and over nuclear magnetic resonance imaging of 54%.
In their review, Morales-Oyarvide et al. (81) reported a sensitivity of 71% using cytology as opposed to a sensitivity of 38% when only using endoscopic ultrasound and concluded that cytologic diagnosis with FNPA is the most appropriate diagnostic test. In our review, we obtained similar data, with a diagnostic sensitivity when using FNPA of 78.5%, a figure similar to that reported by Singhi, Chen, Morales-Oyarvide and Muthusamy (1,79,81,90).
Although the management of cPNETs has generally been surgical, the increased sensitivity and greater use of imaging techniques has led to a rise in the incidental diagnosis of cPNETs, and as a result their treatment has become a matter of controversy especially in elderly patients with a high surgical risk. The therapeutic decision will depend on factors such as potential malignancy, life expectancy, tumor location, the presence of symptoms and the experience of the surgeon. Ligneau et al. (86) proposed that cPNETs should be treated with surgical resection given their possible malignant progression.
Cloyd et al. (77) were the first to classify PNETs depending on their cystic component and concluded that purely cystic PNETs represent a subgroup that may be monitored clinically and radiologically without immediate resection, given their benign progression.
In our review, we found no differences between sexes. The mean age of presentation was 53.2 years (SD = 16.1) but was significantly higher in men (57.1 vs. 49.5; p = 0.001). Of note, 135 cases (44.6%) were diagnosed incidentally, a figure consistent with that from the largest series of solid tumors (3,5). Abdominal pain (35%) was the most frequent symptom. Other phenotypical features of the tumors described are that 87.1% are sporadic and only 7.74% are hereditary and that 85% were non-functional tumors, figures that are higher than those reported for solid tumors (1,2). Another striking characteristic is that the size of non-functional and functional tumors was similar (51 mm vs. 45.8 mm; p = 0.983) when it is well known that in solid tumors non-functional tumors are generally larger. This could be due to the more rapid growth of such tumors because of their hemorrhagic or necrotic component of cPNETs (62,91).
According to the ENETS classification, most tumors correspond to initial stages limited to the pancreas (I, IIa and IIb), with a good prognosis following resection and a 1, 5 and 10 year DFS of 98.5%, 91.5% y 91.5%, respectively. In more advanced stages (IIIb and IV) 1, 5 and 10 year DFS was significantly lower: 100%, 54.2% and 54.2% (p = 0.001) (Fig. 2). The ki-67 index was low in 85.3% of cases and intermediate in 14.9%, figures similar to those reported for solid tumors. These data confirm the prognostic value of the ENETS and WHO classifications for cPNETs (92).
When we analyzed the series according to the degree of functionality, we observed no differences in sex and mean age of presentation. A larger proportion of patients with MEN-1 was observed in the functional tumor group (34.3% vs. 7.3%; p=0.001) and a significantly greater proportion of multifocal tumors (17.6% vs. 2.6%; p=0.016) in the functional tumor group as compared to the non-functional group. When ENETS stage was assessed, we found a higher percentage of functional tumors in stage IV as compared to non-functional tumors (21.2% vs. 3.8%; p = 0.007). DFS was significantly lower in functional tumors (61.9% at 6 years) as compared with non-functional tumors (91.7% at 5 years) (p = 0.001); which is not consistent with the experience reported for solid neuroendocrine tumors (3,93). Perhaps these differences may be explained by the significantly greater proportion of multifocal and MEN-1 tumors in the functional group (p = 0.016 and p = 0.001 respectively).
cPNETs may present as sporadic tumors or in the context of hereditary tumors. The analysis of hereditary cPNETs focused on multiple endocrine neoplasms type 1 (MEN-1) as reports of other entities (Von-Hippel Lindau syndrome or Neurofibromatosis type 1) are anecdotal. Ligneau et al. (86) report that the phenotype of cPNETs associated with MEN-1 syndrome is clinically and pathologically different to that of sporadic cPNETs as they present in younger patients, with a greater percentage of functional tumors which are multiple and generally located in the tail of the pancreas. In our review there were no differences with regard to sex; the mean age of presentation was 41.7 years -lower than in sporadic PNETs due to the high frequency of functional tumors and possible to the better and longer follow-up in patients with this syndrome. The incidence of non-functional cPNET tumors in this subgroup (62.5%) is similar to that reported in other series of patients with MEN-1, although great variability exists in the literature (5,77,94,95). The mean size (42 mm) is not very representative as they are surgical series they reflect the clinical guidelines to treat tumors ≥ 2 cm (5,94).
Disease-free survival in hereditary tumors was lower than that reported for sporadic tumors due to the presence of multifocal tumors and lymph node metastases, a finding in line with that reported by other authors (5,77,95).
We are aware of the limitations of this study such as the great heterogeneity of the data reported in the case reports and in the case reviews and inter-study variability. However, the information reported in case report is more accurate than in large case reviews.
Secondly, this review only includes cases of cPNETs that were resected and histologically confirmed, and thus the real prevalence of cPNETs may not be fully reflected. Furthermore, the cystic morphology seems to be a marker of lower biologic aggressiveness, reducing the number of surgical resections in this type of tumor.
Thirdly, given the size of the sample and the lack of event or loss of follow-up in the patients, it was decided not to undertake a multivariate analysis to determine independent associations.
In contrast to these limitations, case reports and case series are a valuable source of information for evidence-based medicine investigating disorders which are not very prevalent as is our case and which are not subsidiary to randomized studies (6,7,96-99).
To our knowledge, the current review represents the largest number of patients with cPNETs reported in the English literature.
Conclusions
Cystic pancreatic neuroendocrine tumors are an uncommon entity which in 44.5% of cases present incidentally. They represent an entity that is both clinically and pathologically different to solid tumors and must be included in the differential diagnosis of cystic lesions of the pancreas. It seems advisable to follow the same criteria in staging and resectability (≤ 2 cm) in both sporadic and hereditary (MEN-1) tumors. Endoscopic ultrasound in association with FNPA has a high sensitivity and specificity.
Unlike solid tumors, functional cPNETs have a lower overall survival possibly due to the greater incidence of MEN-1 in functional cystic neuroendocrine tumors. In functional cPNETs, the diagnosis of MEN-1 should be ruled out.
In spite of the biologic heterogeneity of cPNETs, the pathologic classification as established by ENETS effectively predicts long-term prognosis in these patients, which confirms the prognostic value of this classification.
References
1. Singhi AD, Chu LC, Tatsas AD, et al. Cystic Pancreatic Neuroendocrine Tumors. Am J Surg Pathol 2012;36(11):1666-73. DOI: 10.1097/PAS.0b013e31826a0048. [ Links ]
2. Gaujoux S, Tang L, Klimstra D, et al. The outcome of resected cystic pancreatic endocrine neoplasms: A case-matched analysis. Surgery 2012;151(4):518-25. DOI: 10.1016/j.surg.2011.09.037. [ Links ]
3. Ellison TA, Wolfgang CL, Shi C, et al. A single institution's 26-year experience with nonfunctional pancreatic neuroendocrine tumors: a validation of current staging systems and a new prognostic nomogram. Ann Surg 2014;259(2):204-12. DOI: 10.1097/SLA.0b013e31828f3174. [ Links ]
4. Birnbaum DJ, Gaujoux S, Cherif R, et al. Sporadic nonfunctioning pancreatic neuroendocrine tumors: prognostic significance of incidental diagnosis. Surgery 2014;155(1):13-21. DOI: 10.1016/j.surg.2013.08.007. [ Links ]
5. Crippa S, Partelli S, Bassi C, et al. Long-term outcomes and prognostic factors in neuroendocrine carcinomas of the pancreas: Morphology matters. Surgery 2016;159(3):862-71. DOI: 10.1016/j.surg.2015.09.012. [ Links ]
6. Moher D, Liberati A, Tetzlaff J, et al. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Int J Surg 2010;8(5):336-41. DOI: 10.1016/j.ijsu.2010.02.007. [ Links ]
7. Gagnier JJ, Kienle G, Altman DG, et al. The CARE guidelines: consensus-based clinical case report guideline development. J Diet Suppl 2013;10(4):381-90. DOI: 10.3109/19390211.2013.830679. [ Links ]
8. Klöppel G, Rindi G, Perren A, et al. The ENETS and AJCC/UICC TNM classifications of the neuroendocrine tumors of the gastrointestinal tract and the pancreas: a statement. Virchows Arch 2010;456(6):595-7. DOI: 10.1007/s00428-010-0924-6. [ Links ]
9. Ricci C, Casadei R, Taffurelli G, et al. Validation of the 2010 WHO classification and a new prognostic proposal: A single centre retrospective study of well-differentiated pancreatic neuroendocrine tumours. Pancreatol Off J 2016;16(3):403-10. DOI: 10.1016/j.pan.2016.02.002. [ Links ]
10. Pergolini I, Iliopoulos O, Ferrone CR, et al. Clinico-pathological features and survival of resected pancreatic neuroendocrine tumors associated with von Hippel Lindau disease and multiple endocrine neoplasia type 1. Gastroenterology 2016;150(4):S1232-3. DOI: 10.1016/S0016-5085(16)34168-3. [ Links ]
11. Amagai H, Jingu K, Kitabayashi H, et al. A case of nonfunctioning neuroendocrine tumor of the pancreas which had difficulty in differentiation with mucinous cystic tumor. Chiba Med J 2016;92(2):45-50. [ Links ]
12. Kawai G, Shirota G, Unno T WT. A case of the NET of the pancreas that was difficult to distinguish from SPT. Japanese J Clin Radiol 2016;61(6):777-81. [ Links ]
13. Tsujino T, Samarasena J CK. Endoscopic ultrasound-guided, needle-based confocal laser endomicroscopy in the diagnosis of cystic pancreatic neuroendocrine tumor: Emerging imaging criteria from surgical pathologic correlation in two cases. Am J Gastroenterol 2015;110(1):S107-8. [ Links ]
14. Lico V, Milanetto AC, Moletta L, et al. Pancreatic neuroendocrine tumor and ileal carcinoid i nacromegaly. Pluriglandular association in non-MEN1 patient: a case report. Neuroendocrinology 2015;102(1-2):151. [ Links ]
15. Humphrey PE, Alessandrino F, Bellizzi AM, et al. Non-hyperfunctioning pancreatic endocrine tumors: multimodality imaging features with histopathological correlation. Abdom Imaging 2015;40(7):2398-410. DOI: 10.1007/s00261-015-0458-0. [ Links ]
16. Apodaca-Torrez FR, Oliveira ML, Triviño T, et al. Tumor neuroendocrino quístico no funcionante del páncreas. Una presentación poco usual. Cir Esp 2015;93(1):43-54. [ Links ]
17. Welch A, McGarrity T, Mani H MM. Cystic neuroendocrine tumor of the pancreas: a rare type of pancreatic cystic lesion accurately diagnosed and staged by EUS-FNA. Pract Gastroenterol 2013;37(10):31-4. [ Links ]
18. Bombardieri R, Moavero R, Roberto D, et al. Pancreatic neuroendocrine tumor in a child with a tuberous sclerosis complex 2 (TSC2) mutation. Endocr Pract 2013;19(5):e124-8. DOI: 10.4158/EP13010.CR. [ Links ]
19. Addeo A, Bini R, Viora T, et al. Von Hippel-Lindau and myotonic dystrophy of Steinert along with pancreatic neuroendocrine tumor and renal clear cell carcinomal neoplasm: Case report and review of the literature. Int J Surg Case Rep 2013;4(8):648-50. DOI: 10.1016/j.ijscr.2013.03.004. [ Links ]
20. Deshmukh SD, Gulati HK, Gaopande V, et al. Incidental cystic endocrine tumor of the pancreas: a case report with immunohistochemical study. J Cancer Res Ther 2012;8(2):289-91. DOI: 10.4103/0973-1482.98992. [ Links ]
21. Hu K-C, Chang W-H, Chu C-H, et al. Pancreatic head well-differentiated endocrine tumor. Am J Surg 2011;201(3):e26-8. DOI: 10.1016/j.amjsurg.2010.03.019. [ Links ]
22. Potru R, Collins C S V. Rare presentation of cystic nonfunctioning pancreatic neuroendocrine tumor: a case-series. Am J Gastroenterol 2011;106(2):S219-20. [ Links ]
23. Baig M, Chin U, Wilhite D CH. Cystic pancreatic neuroendocrine tumors. Am J Gastroenterol 2011;106(2):S213. [ Links ]
24. Higashi T, Kawaguchi Y, Tsukune Y, et al. A case report of glucagon-producing multiple pancreatic neuroendocrine tumors that involved pancreatic cystic lesions. Pancreatology 2010;10(1):70. [ Links ]
25. Diehl DL, Blansfield J, Li J. Cystic pancreatic neuroendocrine tumor. Gastrointest Endosc 2010;71(6):1064-5; discussion 1065. DOI: 10.1016/j.gie.2009.12.016. [ Links ]
26. Colovi? RB, Mati? S V, Micev MT, et al. Two synchronous somatostatinomas of the duodenum and pancreatic head in one patient. World J Gastroenterol 2009;15(46):5859-63. DOI: 10.3748/wjg.15.5859. [ Links ]
27. Kobayashi Y, Yamao K, Sawaki A, et al. A case of pancreatic neuroendocrine tumor (NET) with atypical looking which diagnosed by EUS-FNA. Pancreas 2009;38(8):1018. [ Links ]
28. Basu A, Sistla SC IK. Image of the month. Arch Surg 2009;144(1):89. DOI: 10.1001/archsurg.2008.527-a. [ Links ]
29. Kamei K, Yasuda T, Shinzaki W, et al. Cystic nonfunctioning pancreatic endocrine neoplasm presenting communication with main pancreatic duct. Dig Surg 2009;26(1):25-6. DOI: 10.1159/000193328. [ Links ]
30. Charfi S, Marcy M, Bories E, et al. Cystic pancreatic endocrine tumors: an endoscopic ultrasound-guided fine-needle aspiration biopsy study with histologic correlation. Cancer 2009;117(3):203-10. DOI: 10.1002/cncy.20024. [ Links ]
31. Chetty R, El-Shinnawy I. Intraductal pancreatic neuroendocrine tumor. Endocr Pathol 2009;20(4):262-6. DOI: 10.1007/s12022-009-9093-z. [ Links ]
32. Kongkam P, Al-Haddad M, Attasaranya S, et al. EUS and clinical characteristics of cystic pancreatic neuroendocrine tumors. Endoscopy 2008;40(7):602-5. DOI: 10.1055/s-2007-995740. [ Links ]
33. Ballarin R, Masetti M, Losi L, et al. Cystic pancreatic neuroendocrine neoplasms with uncertain malignant potential: report of two cases. Surg Today 2009;39(2):162-7. DOI: 10.1007/s00595-008-3806-7. [ Links ]
34. Sumarac-Dumanovic M, Micic D, Krstic M, et al. Pitfalls in diagnosing a small cystic insulinoma: a case report. J Med Case Rep 2007;1(1):181. DOI: 10.1186/1752-1947-1-181. [ Links ]
35. Kajiwara M, Gotohda N, Konishi M, et al. Cystic endocrine tumor of the pancreas with an atypical multilocular appearance. J Hepatobiliary Pancreat Surg 2007;14(6):586-9. DOI: 10.1007/s00534-006-1218-x. [ Links ]
36. Li Destri G, Reggio E, Veroux M, et al. A rare cystic non-functioning neuroendocrine pancreatic tumor with an unusual presentation. Tumori 92(3):260-3. [ Links ]
37. Goh BKP, Ooi LLPJ, Tan YM, et al. Clinico-pathological features of cystic pancreatic endocrine neoplasms and a comparison with their solid counterparts. Eur J Surg Oncol 2006;32(5):553-6. DOI: 10.1016/j.ejso.2006.02.017. [ Links ]
38. Tamagno G, Maffei P, Pasquali C, et al. Clinical and diagnostic aspects of cystic insulinoma. Scand J Gastroenterol 2005;40(12):1497-501. DOI: 10.1080/00365520510024160. [ Links ]
39. Imaoka H, Yamao K, Salem AAS, et al. Pancreatic endocrine neoplasm can mimic serous cystadenoma. Int J Gastrointest Cancer 2005;35(3):217-20. DOI: 10.1385/IJGC:35:3:217. [ Links ]
40. Gerke H, Byrne MF, Xie HB, et al. A wolf in sheep's clothing: a non-functioning islet cell tumor of the pancreas masquerading as a microcystic (serous cystic) adenoma. JOP 2004;5(4):225-30. [ Links ]
41. Christein JD MK. Pancreatic somatostitoma associated with obstructive jaundice, pancreatic and biliary Kaposi's sarcoma in a man with neurofibromatosis. HPB 2003;5(1):49-53. DOI: 10.1080/13651820310003575. [ Links ]
42. Vandecaveye V, Verswijvel G, Colla P, et al. Cystic insulinoma of the pancreas in a patient with myotonic dystrophy: correlation of imaging and pathologic findings. JBR-BTR 2003;86(5):268-71. [ Links ]
43. Ahrendt SA, Komorowski RA, Demeure MJ, et al. Cystic pancreatic neuroendocrine tumors: is preoperative diagnosis possible? J Gastrointest Surg 2002;6(1):66-74. [ Links ]
44. Oberg KC, Wells K, Seraj IM, et al. ACTH-secreting islet cell tumor of the pancreas presenting as bilateral ovarian tumors and Cushing's syndrome. Int J Gynecol Pathol 2002;21(3):276-80. DOI: 10.1097/00004347-200207000-00012. [ Links ]
45. Kato K, Kondo S, Ambo Y, et al. Nonfunctioning endocrine tumor of the pancreas with extrapancreatic growth and cyst formation: report of a case. Surg Today 2000;30(7):651-4. DOI: 10.1007/s005950070107. [ Links ]
46. Brown K, Kristopaitis T, Yong S, et al. Cystic glucagonoma: A rare variant of an uncommon neuroendocrine pancreas tumor. J Gastrointest Surg 2(6):533-6. [ Links ]
47. Kotoulas C, Panayiotides J, Antiochos C, et al. Huge non-functioning pancreatic cystic neuroendocrine tumour: a case report. Eur J Surg Oncol 1998;24(1):74-6. DOI: 10.1016/S0748-7983(98)80133-9. [ Links ]
48. Sohaib SA, Reznek RH, Healy JC, et al. Cystic islet cell tumors of the pancreas. AJR Am J Roentgenol 1998;170(1):217. DOI: 10.2214/ajr.170.1.9423637. [ Links ]
49. Sarui H, Yoshimoto K, Okumura S, et al. Cystic glucagonoma with loss of heterozygosity on chromosome 11 in multiple endocrine neoplasia type 1. Clin Endocrinol (Oxf) 1997;46(4):511-6. DOI: 10.1046/j.1365-2265.1997.1380965.x. [ Links ]
50. Zanow J, Gellert K, Benhidjeb T MJ. Cystische tumoren des pankreas. Chirurg 1996;67:719-24. [ Links ]
51. Goto M, Nakano I, Sumi K, et al. Cystic insulinoma and nonfunctioning islet cell tumor in multiple endocrine neoplasia type 1. Pancreas 1994;9(3):393-5. DOI: 10.1097/00006676-199405000-00020. [ Links ]
52. Weissmann D, Lewandrowski K, Godine J, et al. Pancreatic cystic islet-cell tumors. Clinical and pathologic features in two cases with cyst fluid analysis. Int J Pancreatol 1994;15(1):75-9. [ Links ]
53. Iacono C, Serio G, Fugazzola C, et al. Cystic islet cell tumors of the pancreas. A clinico-pathological report of two nonfunctioning cases and review of the literature. Int J Pancreatol 1992;11(3):199-208. DOI: 10.1007/BF02924187. [ Links ]
54. Yagihashi S, Yagihashi N, Nagai K. Cystic Pancreatic Glucagonoma in Contact with Insulinoma Found in a Hypoglycemic Patient. Pathol - Res Pract 1992;188(6):751-6. DOI: 10.1016/S0344-0338(11)80173-1. [ Links ]
55. Nojima T, Kojima T, Kato H, et al. Cystic endocrine tumor of the pancreas. Int J Pancreatol 1991;10(1):65-72. [ Links ]
56. Azoulay D, Sauvanet A, Bonnichon P, et al. Non-functioning tumors of the endocrine pancreas. Concept, diagnosis, treatment. J Chir (Paris) 1990;127(4):185-90. [ Links ]
57. Warshaw AL, Compton CC, Lewandrowski K, et al. Cystic tumors of the pancreas. New clinical, radiologic, and pathologic observations in 67 patients. Ann Surg 1990;212(4):432-43-5. DOI: 10.1097/00000658-199010000-00006. [ Links ]
58. von Herbay A, Sieg B, Otto HF. Solid-cystic tumour of the pancreas. An endocrine neoplasm? Virchows Arch A Pathol Anat Histopathol 1990;416(6):535-8. [ Links ]
59. Davtyan H, Nieberg R, Reber HA. Pancreatic cystic endocrine neoplasms. Pancreas 1990;5(2):230-3. DOI: 10.1097/00006676-199003000-00017. [ Links ]
60. Lapeyrie H, Loizon P, Chapuis H, et al. Non-functional or silent endocrine tumors of the pancreas. Apropos of a case of cystic form. Ann Chir 1989;43(4):302-5. [ Links ]
61. Yagihashi S, Sato I, Kaimori M, et al. Papillary and cystic tumor of the pancreas. Two cases indistinguishable from islet cell tumor. Cancer 1988;61(6):1241-7. DOI: 10.1002/1097-0142(19880315)61:6<1241::AID-CNCR2820610631>3.0.CO;2-T. [ Links ]
62. Kamisawa T, Fukayama M, Koike M, et al. A case of malignant cystic endocrine tumor of the pancreas. Am J Gastroenterol 1987;82(1):86-9. [ Links ]
63. Stair JM, Schaefer RF, McCowan TC, et al. Cystic islet cell tumor of the pancreas. J Surg Oncol 1986;32(1):46-9. DOI: 10.1002/jso.2930320113. [ Links ]
64. Loiterman DA, Schwartz IS, Aufses AH. Cystic islet cell tumor: diagnosis and surgical management. Mt Sinai J Med 1985;52(3):212-5. [ Links ]
65. Heidrich W, Kubale R, Seitz G, et al. Endocrine tumor of the pancreas--a case report on the differential diagnosis of cystic epigastric tumors. Ultraschall Med 1986;7(3):138-40. DOI: 10.1055/s-2007-1011932. [ Links ]
66. Pogany AC, Kerlan RK, Karam JH, et al. Cystic insulinoma. AJR Am J Roentgenol 1984;142(5):951-2. DOI: 10.2214/ajr.142.5.951. [ Links ]
67. Ho PW, Moore GW, Hoge AF. Glucagonoma occurring as a large cystic abdominal mass. South Med J 1984;77(5):666. DOI: 10.1097/00007611-198405000-00036. [ Links ]
68. Thompson NW, Eckhauser FE, Vinik AI, et al. Cystic neuroendocrine neoplasms of the pancreas and liver. Ann Surg 1984;199(2):158-64. DOI: 10.1097/00000658-198402000-00004. [ Links ]
69. Grosdidier J, Duprez J, Delfosse J, et al. Nonfunctional benign Langerhans tumor of the cystic type. Sem Hop 1983;59(23):1773-6. [ Links ]
70. Frija J, Schmit P, Vadrot D, et al. Non-secreting islet cell adenoma of the pancreas evaluated by computed tomography and sonography. Report of a case. Eur J Radiol 1982;2(2):160-1. [ Links ]
71. Caplan RH, Koob L, Abellera RM, et al. Cure of acromegaly by operative removal of an islet cell tumor of the pancreas. Am J Med 1978;64(5):874-82. DOI: 10.1016/0002-9343(78)90531-4. [ Links ]
72. Riddle MC, Golper TA, Fletcher WS, et al. Glucagonoma syndrome in a 19-year-old woman. West J Med 1978;129(1):68-72. [ Links ]
73. Deltz VE PM. Zystisches Insulom. Zbl Chir 10975;100:1521-4. [ Links ]
74. Winston JH. Malignant islet cell adenoma in a pancreatic cyst. Report of a case. J Natl Med Assoc 1965;57(3):203-4. [ Links ]
75. Leger L, Canivet J, Mizon JP, et al. Cyst-like tumors of the islands of Langerhans. Apropos of 2 cases. J Chir (Paris) 1967;94(1):7-20. [ Links ]
76. Joshi RA, Probstein JG. Benign cystic islet-cell tumor of the head of the pancreas; report of a case probably of delta-cell origin. AMA Arch Surg 1955;71(1):74-7. DOI: 10.1001/archsurg.1955.01270130076012. [ Links ]
77. Cloyd JM, Kopecky KE, Norton JA, et al. Neuroendocrine tumors of the pancreas: Degree of cystic component predicts prognosis. Surgery 2016;160(3):708-13. DOI: 10.1016/j.surg.2016.04.005. [ Links ]
78. Mitra V, Nayar MK, Leeds JS, et al. Diagnostic performance of endoscopic ultrasound (EUS)/endoscopic ultrasound--fine needle aspiration (EUS-FNA) cytology in solid and cystic pancreatic neuroendocrine tumours. J Gastrointestin Liver Dis 2015;24(1):69-75. [ Links ]
79. Chen L, Nassar A, Kommineni VT, et al. Endoscopic ultrasonography-guided fine-needle aspiration cytology of surgically confirmed cystic pancreatic neuroendocrine tumors: a Mayo Clinic experience. J Am Soc Cytopathol 2015;4(6):335-43. DOI: 10.1016/j.jasc.2015.04.001. [ Links ]
80. Komminemi VT, Chen L, Zhang J, et al. Cystic pancreatic neuroendocrine tumors: Clinical and endoscopic ultrasound characteristics and accuracy of EUS-FNA. Am J Gastroenterol 2014;109(2):S92. [ Links ]
81. Morales-Oyarvide V, Yoon WJ, Ingkakul T, et al. Cystic pancreatic neuroendocrine tumors: The value of cytology in preoperative diagnosis. Cancer Cytopathol 2014;122(6):435-44. DOI: 10.1002/cncy.21403. [ Links ]
82. Ho HC, Eloubeidi MA, Siddiqui UD, et al. Endosonographic and cyst fluid characteristics of cystic pancreatic neuroendocrine tumours: a multicentre case series. Dig Liver Dis 2013;45(9):750-3. DOI: 10.1016/j.dld.2013.02.021. [ Links ]
83. Konukiewitz B, Enosawa T, Klöppel G. Glucagon expression in cystic pancreatic neuroendocrine neoplasms: an immunohistochemical analysis. Virchows Arch 2011;458(1):47-53. DOI: 10.1007/s00428-010-0985-6. [ Links ]
84. Boninsegna L, Partelli S, D'Innocenzio MM, et al. Pancreatic Cystic Endocrine Tumors: A Different Morphological Entity Associated with a Less Aggressive Behavior. Neuroendocrinology 2010;92(4):246-51. DOI: 10.1159/000318771. [ Links ]
85. Baker MS, Knuth JL, DeWitt J, et al. Pancreatic cystic neuroendocrine tumors: preoperative diagnosis with endoscopic ultrasound and fine-needle immunocytology. J Gastrointest Surg 2008;12(3):450-6. DOI: 10.1007/s11605-007-0219-7. [ Links ]
86. Ligneau B, Lombard-Bohas C, Partensky C, et al. Cystic endocrine tumors of the pancreas: clinical, radiologic, and histopathologic features in 13 cases. Am J Surg Pathol 2001;25(6):752-60. DOI: 10.1097/00000478-200106000-00006. [ Links ]
87. Buetow PC, Parrino T V, Buck JL, et al. Islet cell tumors of the pancreas: pathologic-imaging correlation among size, necrosis and cysts, calcification, malignant behavior, and functional status. AJR Am J Roentgenol 1995;165(5):1175-9. DOI: 10.2214/ajr.165.5.7572498. [ Links ]
88. Bordeianou L, Vagefi PA, Sahani D, et al. Cystic Pancreatic Endocrine Neoplasms: A Distinct Tumor Type? J Am Coll Surg 2008;206(6):1154-8. DOI: 10.1016/j.jamcollsurg.2007.12.040. [ Links ]
89. Yang M, Ke NW, Zeng L, et al. Survival Analyses for Patients With Surgically Resected Pancreatic Neuroendocrine Tumors by World Health Organization 2010 Grading Classifications and American Joint Committee on Cancer 2010 Staging Systems. Medicine (Baltimore) 2015;94(48):e2156. DOI: 10.1097/MD.0000000000002156. [ Links ]
90. Muthusamy VR, Chandrasekhara V, Acosta RD, et al. The role of endoscopy in the diagnosis and treatment of cystic pancreatic neoplasms. Gastrointest Endosc 2016;84(1):1-9. DOI: 10.1016/j.gie.2016.04.014. [ Links ]
91. Iacono C, Verlato G, Ruzzenente A, et al. Systematic review of central pancreatectomy and meta-analysis of central versus distal pancreatectomy. Br J Surg 2013;100(7):873-85. DOI: 10.1002/bjs.9136. [ Links ]
92. Koh YX, Chok AY, Zheng HL, et al. A systematic review and meta-analysis of the clinicopathologic characteristics of cystic versus solid pancreatic neuroendocrine neoplasms. Surgery 2014;156(1):83-96.e2. DOI: 10.1016/j.surg.2014.03.026. [ Links ]
93. Cienfuegos JA, Rotellar F, Salguero J, et al. A single institutions 21-year experience with surgically resected pancreatic neuroendocrine tumors: an analysis of survival and prognostic factors. Rev Española Enfermedades Dig 2016;108(11):689-96. DOI: 10.17235/reed.2016.4323/2016. [ Links ]
94. Thakker R V, Newey PJ, Walls G V, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012;97(9):2990-3011. DOI: 10.1210/jc.2012-1230. [ Links ]
95. Kouvaraki MA, Shapiro SE, Cote GJ, et al. Management of pancreatic endocrine tumors in multiple endocrine neoplasia type 1. World J Surg 2006;30(5):643-53. DOI: 10.1007/s00268-006-0360-y. [ Links ]
96. Vandenbroucke JP. In defense of case reports. Ann Intern Med 2001;134(2 Pt 2):330-4. DOI: 10.7326/0003-4819-134-4-200102200-00017. [ Links ]
97. Agha RA, Fowler AJ, Lee S-Y, et al. Systematic review of the methodological and reporting quality of case series in surgery. Br J Surg 2016;103(10):1253-8. DOI: 10.1002/bjs.10235. [ Links ]
98. Barkin JA, Barkin JS. Pancreatic cysts. Controversies, advances, diagnoses, and therapies. Pancreas 2017;46(6).735-741 DOI: 10.1097/MPA.0000000000000831. [ Links ]
99. Moris M, Wallace MB. Intraductal papillary mucinous neoplasms and mucinous cystadenomas: current status and recommendations. Rev Esp Enferm Dig 2017;109(5):358-67. DOI: 10.17235/reed.2017. 4630/2016. [ Links ]
![]() Correspondence:
Correspondence:
Javier A. Cienfuegos.
Department of General Surgery.
Clínica Universidad de Navarra.
University of Navarra.
Av. Pío XII, 36.
31008 Pamplona, Spain
e-mail: fjacien@unav.es
Received: 08-05-2017
Accepted: 07-08-2017
