










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
Permalink¿CUÁL ES SU DIAGNÓSTICO?
Paciente exfumador, bebedor de 30 g de etanol/día, hipertenso, dislipémico, con cardiopatía isquémica (portador de stent), esteatosis hepática grado IV y quistes renales izquierdos, que acude con carácter urgente al Servicio de Cirugía Oral y Maxilofacial en octubre de 2013 por presentar una tumoración frontal de 1 año de evolución y de crecimiento lentamente progresivo. No refiere otra clínica acompañante salvo visión borrosa ocasionalmente en el ojo derecho, que recupera espontáneamente.
En la tomografía computarizada (TC) craneal (Figura 1) se evidencia una masa sólida que asienta en las partes blandas de la región frontoetmoidal, de predominio derecho, con invasión de los senos frontales y parte anterior de la lámina cribosa; aparentemente no existe invasión de la órbita ni de la fosa craneal anterior. Se identifica otra masa sólida intraorbitaria de menor tamaño (1914 mm), que asienta en el margen superior y lateral de la órbita derecha con adelgazamiento/erosión de la pared ósea orbitaria adyacente. Dada su naturaleza agresiva, son sugestivas de lesiones de origen neoplásico, primario o metastásico. Se solicita una punción-aspiración con aguja fina (PAAF) y una biopsia de la lesión frontal (Figura 2). La analítica, con hemograma y bioquímica, y la tomografía por emisión de positrones-tomografía computarizada (PET-TC) son normales.
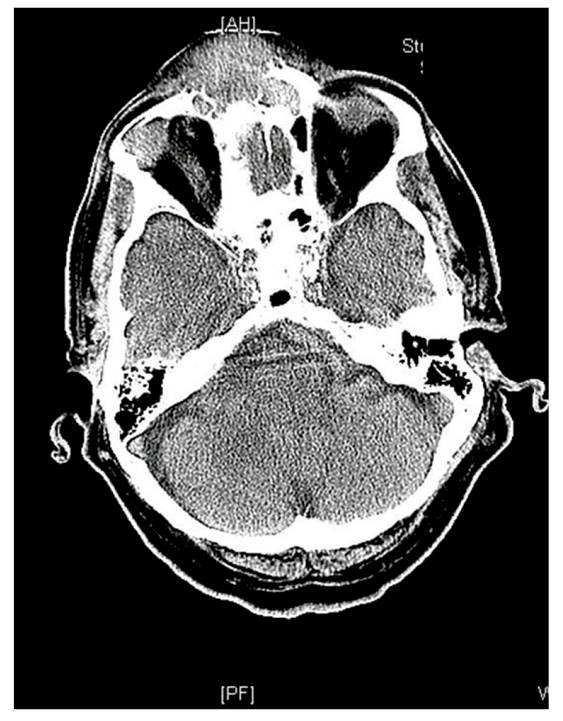
TUMORACIÓN FRONTAL
La PAAF y la biopsia de la lesión frontal describen una neoplasia de células plasmáticas productora de cadenas ligeras kappa y cadenas pesadas gamma (Figura 2). La analítica con hemograma, bioquímica, la ratio de cadenas ligeras libres en suero y la PET-TC son normales. La biopsia de la médula ósea (BMO) presenta un 4,4 % de células plasmáticas clonales, sin sintomatología CRAB (hipercalcemia, fallo renal, anemia y lesiones óseas) típica del mieloma múltiple y banda monoclonal IgG kappa de 1300 mg/dl.

Figura 2. Biopsia de la lesión frontal. Técnica de inmunohistoquímica al microscopio óptico 40x, en la que se observa una neoplasia de células plasmáticas con positividad para cadenas ligeras kappa (imagen), como también así lo fue frente a cadenas pesadas gamma.
El diagnóstico de un plasmocitoma solitario con mínima implicación de la médula ósea (PSMIMO) requiere cumplir los criterios de plasmocitoma solitario elaborados por el Grupo de Trabajo Internacional sobre Mieloma1 (2014) y, además, que las células plasmáticas clonales en la médula ósea sean inferiores al 10 %. En el paciente que nos ocupa la BMO presentaba un 4,4 % de células plasmáticas clonales, por lo que se estableció el diagnóstico de PSMIMO. El paciente recibió tratamiento con radioterapia para el control local de su enfermedad. Debido al aumento de la paraproteína (su persistencia es signo de enfermedad subclínica o diseminación) y la no disminución del tamaño del plasmocitoma frontal con la radioterapia, recibió un ciclo de bortezomib, melfalán y prednisona (no pudo recibir más ciclos por presentar un síndrome coronario agudo sin elevación del ST, tipo angina inestable, que requirió el implante de dos stents farmacoactivos, e infarto agudo de miocardio pericateterismo). No recibió tratamiento desde noviembre de 2014 por disminución franca del plasmocitoma frontal y la paraproteína. El 15 de septiembre de 2015 la TC facial mostraba una clara disminución de la tumoración frontal, pero la serie ósea evidenciaba múltiples pequeñas lesiones osteolíticas en la calota craneal (Figura 3), fémures, ambos húmeros, clavículas, escápulas, parrillas costales y cuerpos vertebrales. Estas lesiones osteolíticas junto con el plasmocitoma frontal son ya diagnósticas de mieloma múltiple, por lo que se retoma el tratamiento anterior y se añade ácido zoledrónico y calcio. El estudio de la médula ósea en diciembre de 2015 no mostraba plasmocitosis en el aspirado, tampoco restricción de inmunoglobulinas en la BMO ni células plasmáticas aberrantes por citometría de flujo.

Figura 3. Radiografía lateral de cráneo. Flechas: múltiples pequeñas lesiones osteolíticas en la calota craneal.
El 3 de agosto de 2016 la TC toracoabdominal y facial seguía mostrando las lesiones líticas previas y objetivaba una masa que ocupaba casi totalmente el antro maxilar derecho y erosionaba y destruía el suelo, proyectándose en el espacio masticador y en la arcada dentaria (Figura 4), además de una tumoración parotídea. El plasmocitoma frontal permanecía sin cambios. Se realizó una biopsia de la lesión maxilar y una PAAF de la tumoración parotídea, que fueron informadas como plasmocitoma (al igual que la tumoración frontal inicial). En esos momentos el estudio de la médula ósea sin biopsia mostraba un 0,6 % de células plasmáticas por citología y un 36 % con fenotipo aberrante por citometría. Ante la aparición de nuevos plasmocitomas se modificó el tratamiento y se pautaron cuatro ciclos de lenalidomida y dexametasona. Actualmente el paciente se encuentra asintomático y con una clara disminución de los plasmocitomas faciales que presentaba (Figura 5).
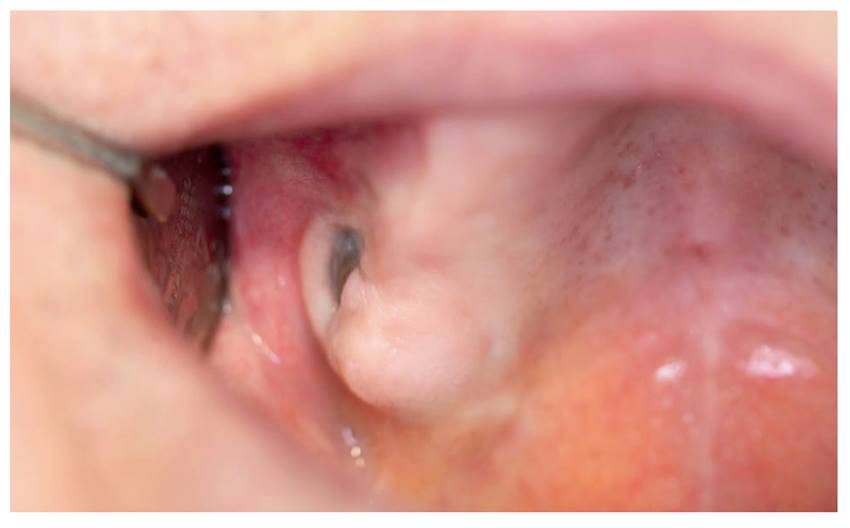
Figura 4. Tumoración blanda con zonas de aspecto ulcerado-ampolloso en la región crestal y vestibular del primer cuadrante.
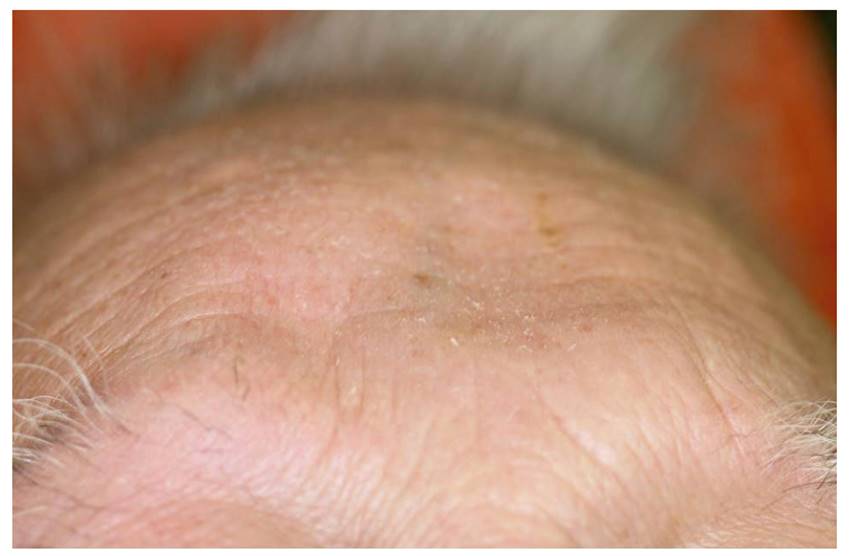
Figura 5. Disminución franca del tamaño de los plasmocitomas frontales con el tratamiento quimioterápico.
El Grupo de Trabajo Internacional sobre Mieloma1 (2014) clasifica las gammapatías monoclonales de significado incierto (GMSI) y otros trastornos relacionados con las células plasmáticas en nueve entidades claramente diferenciadas (Tabla 1). De acuerdo con ello, para el diagnóstico de un plasmocitoma solitario se deben reunir los siguientes datos diagnósticos:
- Biopsia de lesión en tejido óseo o blando con evidencia de células plasmáticas clonales.
- Médula ósea normal sin evidencia de células plasmáticas clonales.
- Estudio normal del esqueleto y resonancia magnética (o TC) de la columna vertebral y la pelvis (excepto la lesión solitaria primaria).
- Ausencia de lesión en ciertos órganos (sintomatología CRAB) que pueda ser atribuida a un trastorno proliferativo de células linfoplasmáticas.
Tabla I. Clasificación de las gammapatías monoclonales de significado incierto (GMSI) y otros trastornos relacionados con las células plasmáticas elaborada por el Grupo de Trabajo Internacional sobre Mieloma1

El plasmocitoma es una neoplasia de células plasmáticas monoclonal que está en la etapa final de maduración de linfocitos B. Esta tumoración asienta en el 90 % de los casos en el hueso (plasmocitoma óseo solitario, POS) y en un 10 % en los tejidos blandos (plasmocitoma extramedular, PEM), como en nuestro caso clínico. Puede ser un tumor aislado, localizarse en cualquier región del cuerpo o ser la primera manifestación de un mieloma múltiple posterior2 3-4.
El PEM representa un 3 % de todos los tumores de células plasmáticas, mientras que el POS representa un 5-10 %. El PEM es más común en hombres, entre la sexta y octava década de la vida. Más del 95 % de los casos ocurren en personas mayores de 40 años. Su incidencia es de 3,5/100.000 al año. Las lesiones extramedulares pueden ocurrir en ausencia de afectación ósea, especialmente en la región de cabeza y cuello. Aproximadamente el 90 % de los PEM se encuentran en cabeza y cuello (suponiendo menos del 1 % de las neoplasias de esta región), y comúnmente implican la cavidad nasal y los senos paranasales3,5).
La etiología de esta enfermedad todavía es desconocida, pero se sugiere la exposición a ciertos productos químicos, los virus, el exceso de irradiación, la estimulación crónica y trastornos genéticos en el sistema reticuloendotelial. En la mayoría de los casos tienen antecedentes de enfermedad autoinmune6.
Los síntomas sistémicos incluyen dolor óseo, fracturas patológicas, insuficiencia renal, hipercalcemia, pérdida de peso, anemia, trombocitopenia y neutropenia. Clínicamente, los PEM se puede manifestar como una masa facial, con aumento de volumen y dolor, obstrucción de la vía aérea, epistaxis, rinorrea, proptosis, disfagia o disfonía. Generalmente aparecen como una masa tumoral, normalmente única, de superficie lisa, subepitelial y de color rosado, gris o rojizo. Pueden ser únicos o múltiples, polipoideos, pediculados o difusos, e incluso a veces ulcerados. La extensión ganglionar regional varía entre un 8 y un 32 %, y solo algunos pacientes desarrollan metástasis a distancia7. El dolor está comúnmente ausente, a menos que haya infección o destrucción ósea. La imagen radiológica varía desde lesiones uniloculares o multiloculares, bien o mal definidas, con bordes nítidos o irregulares, hasta la reabsorción radicular o la reacción perióstica "en sol naciente".
Ocasionalmente presentan gammapatía monoclonal leve, sérica o urinaria (menos de 3 mg/dl), que desaparece tras el tratamiento (su persistencia es signo de enfermedad subclínica o diseminación).
El plasmocitoma es clínicamente similar a ciertas enfermedades inflamatorias crónicas, gingivitis de células plasmáticas y linfomas, con distinta histología. Su diagnóstico diferencial debe considerar el tumor de células mieloides extramedular, el carcinoma indiferenciado, el neuroblastoma olfatorio, el melanoma, el linfoma de células grandes, la plasmocitosis reactiva, el fibrosarcoma, el rabdomiosarcoma, el sarcoma de Ewing, el histiocitoma, el tumor neuroectodérmico periférico y el quiste odontogénico5,8.
El ratio de progresión hacia mieloma múltiple a los 3 años es de un 10 % en el plasmocitoma solitario, un 20 % en el PSMIMO extramedular y un 60 % en el PSMIMO óseo. Los PEM son altamente radiosensibles. La radioterapia se administra para reducir los plasmocitomas en zonas localizadas, consiguiendo el control de la enfermedad local hasta en el 80 % de los casos. La mayoría de los estudios concluyen que una dosis de unos 46 Gy administrados en 4-6 semanas es la mejor opción para el control local, con mínima toxicidad. Por razones estéticas, debe evitarse la cirugía en el manejo de estas lesiones en cabeza y cuello. Cuando sea accesible, la extirpación quirúrgica puede ser considerada en otras áreas y para tratar o prevenir fracturas patológicas. Si hay márgenes quirúrgicos afectados, el paciente debe recibir radioterapia adyuvante3,5. En los pacientes que no responden a la radioterapia o con alto riesgo de progresión, se puede considerar la misma quimioterapia empleada en el mieloma múltiple2,9.
El tratamiento del mieloma múltiple se realiza con quimioterapia, radioterapia y trasplante autógeno de progenitores hematopoyéticos. Los agentes más usados son el melfalán, el bortezomib, la lenalidomida, la talidomida, la ciclofosfamida y la doxorrubicina. Se suelen combinar con prednisona o dexametasona2. Los bisfosfonatos por vía intravenosa reducen la incidencia de fracturas en pacientes con osteopenia o lesiones osteolíticas. Sin embargo, la osteonecrosis mandibular es una complicación potencialmente grave9.
Una de las mayores dificultades en el mieloma múltiple es que, a diferencia de otras enfermedades malignas, la definición de enfermedad es clinicopatológica. Su diagnóstico precisa la presencia de manifestaciones clínicas que implican un serio daño en órganos finales como lesiones osteolíticas y fallo renal. Esta circunstancia ha propiciado que los pacientes no puedan beneficiarse de un tratamiento temprano para prevenir el daño de ciertos órganos, y ha evitado intentos de tratamiento del cáncer en una etapa en que es más susceptible por el estado dependiente del microambiente. Estos criterios eran aceptables en una época donde las opciones de tratamiento eran escasas y tenían efectos tóxicos importantes, y en la que una intervención temprana no había mostrado ningún beneficio clínico aparente. Sin embargo, esta actitud ya no puede estar justificada, debido a que las opciones de tratamiento han mejorado notablemente y los datos muestran que una intervención temprana en los pacientes asintomáticos de alto riesgo puede prolongar la supervivencia1. Hay que destacar, en el caso que nos ocupa, que la presencia de un POS o un PEM demostrada mediante biopsia y la existencia de una o más lesiones osteolíticas en la radiografía esquelética (TC o PET-TC) son ya suficientes para el diagnóstico de mieloma múltiple. Y con los nuevos criterios diagnósticos de 2014, esa misma biopsia junto con más de una lesión focal de 5 mm o más en un estudio de resonancia magnética, también lo es.
Por todo lo anteriormente expuesto, el cirujano oral y maxilofacial debe incluir esta posibilidad en el diagnóstico diferencial de lesiones radiolúcidas, no demorar su estudio y derivar al paciente para facilitar un tratamiento precoz, teniendo presente la posible transformación de un plasmocitoma en un mieloma múltiple, como el caso clínico expuesto, por lo que el seguimiento debe ser estrecho de por vida.

