










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNeurocirugía
versión impresa ISSN 1130-1473
Neurocirugía vol.18 no.5 oct. 2007
Biología molecular de los glioblastomas
Biology molecular of glioblastomas
C. Franco-Hernández; V. Martínez-Glez y J.A. Rey
Unidad de Investigación. Fundación para la Investigación Biomédica del Hospital Universitario La Paz. Madrid.
Dirección para correspondencia
RESUMEN
La formación de los glioblastomas es muy diversa, pudiendo presentarse de "novo" o provenir de recidivas de astrocitomas que van progresando hacia mayores grados de malignidad. La alteración molecular más frecuente que se encuentra en estos tipos tumorales es la pérdida de heterocigocidad del cromosoma 10 en el que se han identificado varios genes supresores de tumores. Las vías genéticas TP53/MDM2/ P14arf y CDK4/RB1/P16ink4 implicadas en división celular, se encuentran desreguladas en la mayoría de los gliomas así como los genes que promueven la división celular, entre ellos EGFR. Por último el aumento de factores de crecimiento y angiogénicos también está involucrado en el desarrollo de estos tipos tumorales. Uno de los objetivos de la biología molecular en tumores de estirpe glial es intentar encontrar marcadores o alteraciones genéticas que permitan abordar mejor la clasificación de los glioblastomas, su evolución y pronóstico así como su tratamiento. La diversidad y la cantidad de las alteraciones moleculares presentes en glioblastomas probablemente sea el motivo por el que todavía no se han encontrado fármacos efectivos para combatirlos. En la actualidad, con la aparición de nuevas técnicas de biología molecular, se puede intentar individualizar y clasificar a los pacientes en función de su expresión génica. Esto abre una ventana esperanzadora a la aparición de nuevos fármacos que tengan como diana exclusiva a los genes y/o proteínas alterados de las células tumorales en función de su patrón de expresión génica individualizado para cada tumor. En este artículo revisamos los mecanismos moleculares más frecuentes en la patogénesis de los glioblastomas.
Palabras clave: Glioblastoma. Biología molecular.
SUMMARY
Glioblastomas, the most frequent and malignant human brain tumors, may develop de novo (primary glioblastoma) or by progression from low-grade or anapalsic astrocytoma (secondary glioblastoma). The molecular alteration most frequent in these tumorlike types is the loss of heterozygosity on chromosome 10, in wich several genes have been identified as tumors suppressor. The TP53/MDM2/P14arf and CDK4/RB1/ P16ink4 genetic pathways involved in cycle control are deregulated in the majority of gliomas as well as genes that promote the cellular division, EGFR. Finally the increase of growth and angiogenics factors is also involved in the development of glioblastomas. One of the objetives of molecular biology in tumors of glial ancestry is to try to find the genetic alterations that allow to approach better the classification of glioblastomas, its evolution prediction and treatment.
The new pathmolecular classification of gliomas should improve the old one, especially being concerned about the oncogenesis and heterogenity of these tumors. It is desirable that this classification had clinical applicability and integrates new molecular findings with some known histological features with pronostic value.
In this paper we review the most frequent molecular mechanisms involved in the patogenesis of glioblastomas.
Introducción
El glioblastoma multiforme es el tumor más frecuente de los diferentes tipos de gliomas que existen y desafortunadamente el más maligno35. Constituye alrededor del 50% de todos los gliomas así como el 25% de todos los tumores intracraneales. Es un tumor difusamente infiltrante y pobremente diferenciado de la células gliales. En la actualidad se considera como el grado de malignidad extremo de continuidad de los tumores astrocíticos. Suele originarse en la sustancia blanca y la denominación multiforme está dada por la heterogeneidad que lo caracteriza bajo visión microscópica51. Puede desarrollarse a partir de un astrocitoma de bajo grado que va sufriendo transformación anaplásica e ir evolucionando hasta un glioblastoma secundario (GBM2); o directamente presentarse como un glioblastoma primario o de "novo"(GBM1).
La biología de los gliomas malignos se asocia con el balance de la expresión de las proteínas que controlan de manera positiva o negativa el ciclo celular, la proliferación, la motilidad, la neoformación vascular y el reconocimiento del sistema inmune. Estos fenómenos son el resultado de cambios en el nivel de expresión de un gen normal o de involucrar la pérdida de su expresión. La señal para que la célula normal prolifere comúnmente se inicia en la superficie celular, donde los factores de crecimiento liberados por la matriz extracelular o por el propio tumor interactúan con los receptores específicos en la membrana, desencadenando diversos mecanismos de señalización intracelular que afectan a la expresión génica para promover la división celular. Pueden existir alteraciones y mutaciones en cada una de estas vías que en conjunto o de forma independiente pueden dar lugar a la pérdida de regulación del ciclo celular, la angiogénesis, la proliferación y/o favorecer la invasión18.
La frecuencia de las alteraciones genéticas que están presentes en GBM2 y GBM1 son diferentes así como la edad de los pacientes en la que se presentan. Mientras que los GBM1 suelen aparecer en edades más tardías, alrededor de los 60-70 años, los GBM2 suelen presentarse en edades más tempranas, 40-50 años. En la génesis del glioblastoma existen alteraciones moleculares a nivel de genes supresores de tumores (GST), oncogenes y genes reparadores de ADN. La figura 1 muestra la progresión de los gliomas en función de sus alteraciones moleculares, que detallamos a continuación.

Figura1. Esquema de los diferentes mecanismos y alteraciones
moleculares más frecuentes implicadas en la génesis de un glioblastoma.
Alteraciones de genes localizados en el cromosoma 10
La alteración más frecuente identificada en glioblastomas es la pérdida de heterocigocidad (LOH) en 10q58. La frecuencia de LOH en 10q es similar en GBM1 (70%) y en GBM2 (63%)50 aunque en GBM1 se ha observado que la pérdida es del cromosoma 10 completo en la mayoría de los casos, mientras que en GBM2 se observa generalmente pérdida de 10q pero no de 10p27. En el cromosoma 10 se han identificado varios GST: PTEN (homólogo de la tensina y fosfatasa) en 10q23.3, DMBT1 (supresor de tumores cerebrales malignos) localizado en 10q25.3-q26.1, FGFR2 (receptor del factor del crecimiento fibroblástico) en la región 10q26 y un gen reparador de ADN, la O6-metilguanina-DNAmetiltransferasa (MGMT), se localiza en 10q2649. Estos GST intervienen en el control del ciclo celular y reparación de ADN. La inactivación de un GST ha de producirse en homocigosis, para que el gen pierda completamente su funcionalidad. En glioblastomas una copia de PTEN, DMBT1, FGFR2 y/o MGMT se pierde normalmente por LOH 10q y la otra copia del gen está sujeta a mutaciones o a procesos epigenéticos que lo inactivan26,30,43.
En glioblastomas se han observado alteraciones epigenéticas, que se definen como aquéllas que influyen en la actividad del gen pero no implican cambios en la secuencia del ADN8. La principal modificación epigenética en humanos es la metilación de la citosina localizada en los dinucleótidos CpG. Existen lugares donde son más abundantes, denominándose islas CpG que normalmente no están metiladas en las células no tumorales20. La metilación aberrante de las islas CpG produce una parada de la transcripción del gen originando su inactivación. Existen muchos GST y genes reparadores del ADN que pierden la expresión mediante este mecanismo en tumores de estirpe glial, así como también en otros tipos de cáncer30.
Las mutaciones de PTEN son más frecuentes en GBM1 (25%) pero no exclusivas, ya que algunos autores han identificado mutaciones de PTEN en GBM2 (4%)55. Se han identificado alrededor de 78 mutaciones diferentes de PTEN en glioblastomas, de las cuales el 12,8% son mutaciones silenciosas, el 32,1% son deleciones o inserciones distribuidas por todos los exones y el 33% son mutaciones con cambio de aminoácido; estas mutaciones se han localizado preferentemente en los exones 1-6 de PTEN, región que presenta homología con ciertos dominios de las fosfatasas. PTEN, como fosfatasa que es, puede retirar el fosfato del fosfatidil inositol 3 fosfato (PIP3), deteniéndose la señal de crecimiento celular y provocando generalmente una disminución de la actividad celular. Dado que la deleción en homocigosis de PTEN es bastante rara36, otro mecanismo que se postula para su inactivación es la metilación de su promotor9. Las mutaciones de PTEN son más frecuentes en glioblastomas que en astrocitomas anaplásicos23. Estos datos sugieren que dichas mutaciones constituyen una alteración importante en el desarrollo de gliomas, pudiendo representar un paso molecular necesario en la transformación de gliomas de bajo a alto grado de malignidad.
Las mutaciones de DMBT1 son bastante escasas en glioblastomas, por lo que la pérdida de función del gen se debe principalmente a una deleción en homocigosis46; esta alteración ya se observa en astrocitomas de bajo grado23. La pérdida de función de DMBT1 se postula que podría estar sujeta a la metilación en su promotor pero todavía no existen suficientes trabajos que lo permitan afirmar de forma concluyente. También se postula que FGFR2 podría funcionar como GST, ya que se pierde su expresión en estos tipos de tumores, pero todavía no se conoce muy bien su función en la génesis de los gliomas26.
El gen MGMT, cuya pérdida de expresión suele estar vinculada a hipermetilación, codifica para una proteína reparadora de ADN que actúa eliminando los radicales mutagénicos y citotóxicos en posición O6 guanina del ADN. Para ello, transfiere estos radicales a una cisterna interna en una reacción que inactiva a una molécula de MGMT por cada lesión reparada. De este modo, la capacidad de reparación de lesiones en el ADN de una célula, depende del número de moléculas de MGMT y por tanto de la tasa de síntesis de novo de la misma22. La acumulación de mutaciones en gliomas subsecuente a la inactivación de MGMT no parece ser aleatoria ya que si bien esta inactivación produce acumulación de mutaciones en el oncosupresor TP53, dichas alteraciones son prácticamente inexistentes en otro gen, TP73, estrechamente relacionado con TP532,3. Esteller y colaboradores demostraron que la metilación aberrante de MGMT producía una mejor respuesta a tratamientos con temozolomida en gliomas21. Por lo tanto la ausencia de la proteína o la inactivación del gen es un factor de mal pronóstico debido a que los pacientes que poseen metilación en este gen acumulan más mutaciones, pero a la vez también podría representar un factor predictivo de respuesta a la quimioterapia en este tipo de tumores.
Alteraciones de la vía TP53/MDM2/P14arf
La vía genética TP53/MDM2/P14arf también está sujeta a alteraciones en glioblastomas y astrocitomas12,37. Es una vía implicada en respuesta a estrés celular que va a provocar la activación de genes relacionados con el control del ciclo celular, reparación de ADN y apoptosis4. TP53 es un gen que codifica para la proteína p53 que actúa como factor de transcripción (FT) uniéndose a los promotores de genes implicados en la reparación de ADN. En condiciones normales p53 está secuestrado por su represor MDM2 por lo que la proteína no es funcional; cuando la célula entra en división (fase G1-S), MDM2 libera a p53. Este FT va a promover la trascripción de otros genes implicados en reparación de ADN y/o apoptosis. En las células que no presentan alteraciones en esta vía y que poseen su material genético dañado, p53 se une a los promotores que van a activar a genes implicados en la reparación del ADN; sin embargo, si el material genético no puede ser reparado la célula entra en apoptosis antes de dividirse. Por contra, si la célula posee mutaciones en p53 los daños producidos en el ADN no podrán ser reparados provocando un aumento de la división celular y una disminución en la apoptosis y en las reparaciones del material genético. En esta vía también interviene p14arf, esta proteína es un represor de MDM2 cuando no esta unida a p53. En caso de que p14arf no ejerza su correcta función, va a desencadenar un exceso de MDM2 libre, como consecuencia MDM2 secuestrará en unos niveles mayores a los normales a p53 y como resultado se producirá una mayor acumulación de daños en el ADN. Esta suma de alteraciones conduce a la célula a desencadenar un proceso tumoral. Alrededor del 65% de GBM2 presentan mutaciones en TP53; esta alteración molecular también está presente en astrocitomas anaplásicos y astrocitomas de bajo grado de malignidad por lo que se postula que sea un evento temprano en la transformación neoplásica50,59. Las mutaciones en TP53 frecuentemente encontradas en GBM2 son las transiciones G:C → A:T. Alrededor del 57% de estas transiciones han sido identificadas en los codones 248 y 273 del gen, representando puntos calientes y van a estar sujetos a una mayor tasa de mutación que el resto de los exones50. Por el contrario, las mutaciones de TP53 en GBM1 son menos abundantes, no sobrepasando el 28%. Las mutaciones de este gen en los GBM1 están distribuidas por todos los exones sin existir puntos preferenciales a sufrir mutación, y en la mayoría de los casos constituyen eventos secundarios debido a la inestabilidad genómica durante la progresión del tumor50.
Por otra parte, se ha observado que alrededor del 10% de GBM1 sufren amplificación de MDM2, cuando TP53 no se encuentra alterado14,52, evento bastante infrecuente en GBM2. Se han identificado deleciones en homozigosis de p14arf en GBM1 y se ha comprobado que alrededor de 1/3 de los astrocitomas de bajo grado presentan metilación en el promotor de p14arf.12,60. Así pues, la inactivación de p14arf suele ser por deleción en homocigosis en GBM1 y por metilación en GBM247.
Mutaciones en TP53, amplificaciones de MDM2 y/o deleciones y metilaciones de p14arf son alteraciones moleculares que dan lugar a un acúmulo de otras mutaciones en el ADN, pudiendo originar una muerte celular o una trasformación neoplásica.
En un principio se creyó que las mutaciones de TP53 eran exclusivas de GBM2 por lo que mediante un análisis mutacional de este gen, seríamos capaces de distinguir entre un GBM1 y un GBM2. En la actualidad varios grupos de investigación también han encontrado mutaciones en GBM1 por lo que esta teoría se ha descartado. Schmidt et al53 formulan que la presencia de mutaciones en TP53 es un factor pronóstico de supervivencia debido a que las mutaciones de este gen ya se encuentran en gliomas de bajo grado, que comportan un mejor pronóstico. Sin embargo, dado que ahora se admite que las mutaciones de TP53 también están presentes en GBM1, la capacidad predictiva de estas alteraciones debería ser revisada.
Alteraciones de la vía p16ink4/RB1/CDK4
Otra de las vía afectadas en glioblastomas es p16ink4/ RB1/CDK432. RB1 (gen del retinoblastoma) se localiza en 13q14.2, su proteína es la que controla la transición G1-S en el ciclo celular54. Cuando rb1 no está fosforilado secuestra a E2F, un factor de trascripción que activa genes implicados en la transición G1-S del ciclo celular cuando no está unido a rb1. La fosforilación de rb1 es producida por CDK4 (ciclina dependiente de la quinasa 4) y la proteína encargada de inhibir a esta ciclina es p16ink4 (inhibidor de la ciclina dependiente de la quinasa 4). La pérdida en homozigosis de p16ink4, y/o la amplificación de CDK4 provoca que rb1 esté continuamente fosforilada y no pueda unirse a E2F; como resultado se produce una división celular incontrolada. Estas dos alteraciones se encuentran presentes en glioblastomas15,32 así como también la metilación del promotor de RB1, aunque esta metilación es más frecuente en GBM2 que en GBM129,48. En astrocitomas de bajo grado, hasta el momento no existen suficientes datos que nos demuestren que existe metilación del promotor de RB1. En astrocitomas anaplásicos es bastante infrecuente a metilación de dicho promotor, por lo que se cree que esta alteración sería un evento tardío en la progresión del astrocitoma a GBM2.
La figura 2 muestra de forma esquemática como están relacionadas la vía TP53/MDM2/P14arf y la vía p16ink4/RB1/ CDK4 en el control del ciclo celular.
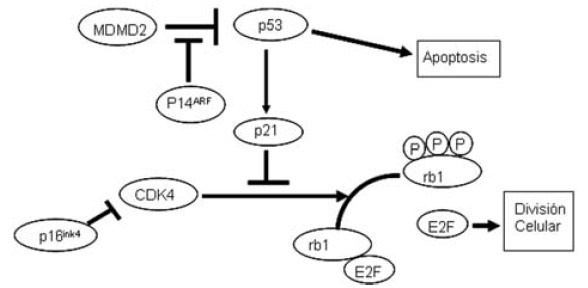
Figura 2. La figura representa la conexión de dos de las vías que controlan el ciclo celular,
TP53/MDM2/P14arf y CDK4/ P14ink4/RB1. estas dos vías se encuentran alteradas en glioblastomas.
La alteración de la vía TP53/MDM2/P14arf conlleva una disminución de la apoptosis sin embargo
la alteración de la vía CDK4/P14ink4/RB1 da lugar directamente a un aumento de la división celular.
Represor Activador
Alteraciones en el gen EGFR
Otra de las alteraciones genéticas presentes en glioblastomas es la amplificación del gen codificador del receptor del factor de crecimiento epidérmico (EGFR)5,33. EGFR se localiza en 7p12 y su sobrexpresión génica puede deberse, entre otras alteraciones, a una amplificación en tándem que da lugar a diferentes variantes de la proteína; la más común es la variante EGFRvIII13. Esta alteración estructural se debe preferentemente a una deleción de 801 pares de bases (p.b.), que abarca desde los exones 2 hasta el 7 y una posterior fusión del resto de los exones, dando lugar a una amplificación en tándem del gen. Ello provoca la síntesis de la proteína con el receptor truncado y constitutivamente activado independientemente del ligando; como resultado la célula comienza a dividirse de forma incontrolada10. La detección de EGFRvIII por inmunohistoquímica no parece estar relacionada con la supervivencia en los GBM pero sí con los astrocitomas anaplásicos, lo cual sugiere que la detección de EGFRvIII puede ser utilizada para identificar y/o confirmar la identidad de un astrocitoma que se comportaría como un GBM1. La habilidad de las células tumorales para generar estas formas mutantes funcionales de EGFR puede contribuir a la capacidad de los gliomas para evadir la quimioterapia, así como las mutaciones presentes en el gen también pueden influir en el pronóstico o el tratamiento a utilizar40,31. La figura 3 ilustra una deleción de 18 p.b. del exon 19 de EGFR en un GBM1.

Figura 3. Deleción de 18 p.b. del exon 19 de EGFR en un GBM1.
Técnica PCR/SSCP y posterior secuenciación automática.
EGFR (Receptor del factor de crecimiento epidérmico),
N = ADN no tumoral, GBM1 = ADN procedente de un
glioblastoma primario. En el ADN no tumoral se observa
únicamente una secuencia, por el contrario el ADN del
glioblastoma primario muestra una doble secuencia debido
a la deleción del 18 p.b. Las flechas indican el inicio y el final
de la deleción.
Otro de los mecanismos por los que puede existir una sobredosis del gen sería la polisomía del cromosoma 7 que frecuentemente se observa en este tipo de tumores41,11. Por último, la amplificación de EGFR ocurre en el 40% de GBM1 siendo abundante en GBM219,61. Inicialmente se pensó que esta alteración era exclusiva de GBM1 con respecto a GBM2 pero con el tiempo otros trabajos han demostrado que en GBM2 también puede existir amplificación de EGFR,19,61.
En la tabla 1 se muestra un resumen de los tipos de alteraciones moleculares y genes alterados que se identifican en gliomas.
Factores de crecimiento y angiogénicos sobrexpresados en glioblastomas
Para el crecimiento celular se requiere un balance entre los factores angiogénicos y antiangiogénicos. La remodelación de los vasos sanguíneos se inicia por las señales de crecimiento que interactúan con su ligando en la superficie celular. La activación no regulada de la angiogénesis puede ser el paso de una neoplasia benigna a una maligna. Esta hipótesis se ha formulado en gliomas de bajo grado que progresan a glioblastoma multiforme. Uno de los principales estímulos para la síntesis de factores angiogénicos es la hipoxia, que induce la síntesis del factor de crecimiento endotelial (VEGF)34,38,57. Se considera que el VEGF es el mediador más importante en la neovascularización de gliomas y su expresión aumenta durante el proceso de malignización de los gliomas42. VEGF tiene tres receptores (VEGFR-1,2,3), dos de ellos, el VEGFR-1 y el VEGFR-3 están sobrexpresados en las células endoteliales de astrocitomas y oligodendrogliomas anaplásicos así como también en los ependimomas16.
Hay otros factores de crecimiento que también juegan un papel importante en la génesis y formación de un glioma. En general estos factores de crecimiento interaccionan con proteínas tirosín-kinasas para inducir la dimerización, autofosforilación y expresión de sitios de unión para que interaccionen las proteínas de señal. El complejo así formado activa una cascada de señales por parte de unas proteínas llamadas transductores de señales y activadores de la trascripción (STATs) que provocará un aumento de la división celular debido a la activación de genes antiapoptóticos y genes que intervienen en la división celular. Los factores de crecimiento que están involucrados en la génesis de un glioma son:
1. EGF (Factor de crecimiento epidérmico): provoca proliferación celular por estimulación autocrina, resistencia a la quimioterapia y aumento de la tumorigenicidad24. Su receptor es EGFR.2. Factor de crecimiento fibroblástico (FGF): provoca proliferación celular por estimulación autocrina y además presenta un efecto sinérgico con VEGF y el factor de crecimiento hepatocítico (HGF) para estimular la angiogénesis7. Se ha descrito que existe incremento de FGF y sus receptores en gliomas45.
3. VEGF: es el principal mediador de la angiogénesis involucrado en el aumento de edema cerebral y también actúa en la proteolisis de las proteínas de la matriz extracelular17.
4. Factor de crecimiento plaquetario (PDGF): potencia a VEGF para estimular la angiogénesis y está involucrado en el aumento de la trascripción de los factores angiogénicos. Suele estar sobrexpresado en gliomas de bajo grado teniendo implicaciones pronósticas56.
5. HGF: cuya actividad provoca un aumento de resistencia a la quimioterapia39, aumento de la proliferación celular6, estimulación de la angiogénesis48. Los gliomas de alto grado de malignidad contienen niveles más elevados de HGF que los gliomas de bajo grado o el tejido sano44.
6. Factor de crecimiento insulínico (IGF): interviene en la proliferación celular por estimulación paracrina28. Se encuentran sobrexpresados en gliomas de bajo grado y otras neoplasias del SNC28,62 relacionándose con un proceso de proliferación y disminución de la apoptosis25.
Perspectivas futuras
Se han realizado grandes esfuerzos para poder inhibir a estos factores de crecimiento e interrumpir sus señales intracelulares que regulan la invasividad, angiogénesis, proliferación y apoptosis. La dificultad para obtener tratamientos satisfactorios quizás radique en que los gliomas malignos presentan alteraciones múltiples y donde se encuentran muchas vías genéticas de señalización y control de la proliferación celular implicadas, mostrando mucha heterogeneidad molecular incluso dentro del mismo tumor. Para intentar combatir el desarrollo de estos tumores con tratamientos satisfactorios, son necesarios tratamientos múltiples para abordar todas las vías que se encuentran alteradas. Ello requiere una caracterización molecular de los gliomas para poder establecer el mejor tratamiento en función de las alteraciones y patrón de expresión génico. En un futuro se esperan identificar patrones de expresión molecular en gliomas y de esta forma poder aproximarnos mejor al pronóstico y al tratamiento. Sin embargo, todavía no es posible la clasificación individual a pesar de la enorme cantidad de esfuerzos que se están realizando. Aun así la investigación de nuevos mecanismos y vías implicadas en la génesis de los gliomas hacen que permanezcamos cuidadosamente optimistas. Para intentar combatir los gliomas en particular y los tumores cerebrales en general, se requiere la estrecha colaboración de los profesionales de las distintas especialidades.
Bibliografía
1. Aldape, K.D., Ballman, K., Furth, A., Buckner, J.C., Giannini, C., Burger, P.C., Scheithauer, B.W., Jenkins, R.B., James, C.D.; Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J Neuropathol Exp Neurol. 2004; 63: 700-707. [ Links ]
2. Alonso, M.E., Bello, M.J., González-Gómez, P., Lomas, J., Arjona, D., de Campos, J.M., Kusak, M.E., Sarasa, J.L., Isla, A., Rey, J.A.: Mutation analysis of the p73 gene in nonastrocytic brain tumours. Br J Cancer. 2001; 85: 204-208. [ Links ]
3. Alonso, M.E., Bello, M.J., Lomas, J., González-Gómez, P., Arjona, D., De Campos, J.M., Gutiérrez, M., Isla, A., Vaquero, J., Rey, J.A.: Absence of mutation of the p73 gene in astrocytic neoplasms. Int J Oncol. 2001 ;19: 609-612. [ Links ]
4. Amundson, S.A., Myers, T.G., Fornace, A.J. Jr.: Roles for p53 in growth arrest and apoptosis: putting on the brakes after genotoxic stress. Oncogene. 1998; 17: 3287-3299. [ Links ]
5. Arjona, D., Bello, M.J., Rey, J.A.: EGFR intragenic loss and gene amplification in astrocytic gliomas. Cancer Genet Cytogenet. 2006; 164: 39-43. [ Links ]
6. Arrieta, O., García, E., Guevara, P., García-Navarrete, R., Ondarza, R., Rembao, D., Sotelo, J.: Hepatocyte Growth Factor Is Associated with Poor Prognosis of Malignant Gliomas and Is a Predictor for Recurrence of Meningioma. Cancer 2002; 94: 3210-3218. [ Links ]
7. Auguste, P., Gursel, D.B., Lemiere, S., Reimers, D., Cuevas, P., Carceller, F., Di Santo, J.P., Bikfalvi, A.: Inhibition of fibroblast growth factor/fibroblast growth factor receptor activity in glioma cells impedes tumor growth by both angio-genesis-dependent and -independent mechanisms. Cancer Res 2001; 61: 1717-1726. [ Links ]
8. Bae, S.I., Lee, H.S., Kim, S.H., Kim, W.H.: Inactivation of O6-methylguanine-DNA methyltransferase by promoter CpG island hypermethylation in gastric cancers. Br J Cancer. 2002; 86: 1888-1892. [ Links ]
9. Baeza, N., Weller, M., Yonekawa, Y., Kleihues, P., Ohgaki, H.: PTEN methylation and expression in glioblastomas. Acta Neuropathol (Berl). 2003; 106: 479-85. [ Links ]
10. Batra, S.K., Castelino-Prabhu, S., Wikstrand, C.J., Zhu, X., Humphrey, P.A., Friedman, H.S., Bigner, D.D.: Epidermal growth factor ligand-independent, unregulated, cell-transforming potential of a naturally occurring human mutant EGFRvIII gene. Cell Growth Differ. 1995; 6: 12511259. [ Links ]
11. Bello, M.J., de Campos, J.M., Kusak, M.E., Vaquero, J., Sarasa, J.L., Petana, A., Rey, J.A.: Ascertainment of chromosome 7 gains in malignant gliomas by cytogenetic and RFLP analyses. Cancer Genet Cytogenet. 1994; 72: 55-58. [ Links ]
12. Bello, M.J., Rey, J.A.: The p53/Mdm2/p14ARF cell cycle control pathway genes may be inactivated by genetic and epigenetic mechanisms in gliomas. Cancer Genet Cytogenet. 2006; 164: 172-173. [ Links ]
13. Biernat, W., Huang, H., Yokoo, H., Kleihues, P., Ohgaki, H.: Predominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomas. Brain Pathol. 2004; 14: 131-136. [ Links ]
14. Biernat, W., Kleihues, P., Yonekawa, Y., Ohgaki, H. Amplification and overexpression of MDM2 in primary (de novo) glioblastomas. J Neuropathol Exp Neurol. 1997; 56: 180-185. [ Links ]
15. Burns, K.L., Ueki, K., Jhung, S.L., Koh, J., Louis, D.N.: Molecular genetic correlates of p16, cdk4, and pRb immunohistochemistry in glioblastomas. J Neuropathol Exp Neurol. 1998; 57: 122-130. [ Links ]
16. Chan, A.S., Leung, S.Y., Wong, M.P., Yuen, S.T., Cheung, N., Fan, Y.W., Chung, L.P.: Expression of vascular endothelial growth factor and its receptors in the anaplastic progression of astrocytoma, oligodendroglioma, and ependymoma. Am J Surg Pathol. 1998; 22: 816-826. [ Links ]
17. Chaudhry, I.H., O'Donovan, D.G., Brenchley, P.E., Reid, H., Roberts, I.S.: Vascular endothelial growth factor expression correlates with tumour grade and vascularity in gliomas. Histopathology. 2001; 39: 409-415. [ Links ]
18. Demuth, T., Berens, M.E.: (2004) Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004; 70: 217-228. [ Links ]
19. Ekstrand, A.J.,Sugawa, N., James, C.D., Collins, V.P.: Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N-and/or C-terminal tails. Proc Natl Acad Sci USA. 1992; 89: 4309-4313. [ Links ]
20. Esteller, M., Fraga, M.F., Paz, M.F., Campo, E., Colomer, D., Novo, F.J., Calasanz, M.J., Galm, O., Guo, M., Benitez, J., Herman, J.G.: Cancer epigenetics and methylation. Science. 2002; 297: 1807-1808. [ Links ]
21. Esteller, M., García-Foncillas, J., Andion, E., Goodman, S.N., Hidalgo, O.F., Vanaclocha, V., Baylin, S.B., Herman, J.G.: Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000; 343: 1350-1354. [ Links ]
22. Esteller, M., Hamilton, S.R., Burger, P.C., Baylin, S.B., Herman, J.G.: Inactivation of the DNA repair gene O6methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999; 59: 793-797. [ Links ]
23. Fan, X., Muñoz, J., Sanko, S.G., Castresana, J.S.: PTEN, DMBT1, and p16 alterations in diffusely infiltrating astrocytomas. Int J Oncol. 2002; 21: 667-674. [ Links ]
24. Frederick, L.: Diversity and frequency of epidermal growth factor receptor mutations in humanglioblastomas. Cancer Res. 2000; 60: 1383-1387. [ Links ]
25. Friend, K.E., Khandwala, H.M., Flyvbjerg, A., Hill, H., Li, J., McCutcheon, I.E.: Growth hormone and insulinlike growth factor-I: effects on the growth of glioma cell lines. Growth Horm IGF Res. 2001; 11: 84-91. [ Links ]
26. Fujisawa, H., Kurrer, M., Reis, R.M., Yonekawa, Y., Kleihues, P., Ohgaki, H.: Acquisition of the glioblastoma phenotype during astrocytoma progression is associated with loss of heterozygosity on 10q25-qter. Am J Pathol. 1999; 155: 387-394. [ Links ]
27. Fujisawa, H., Reis, R.M., Nakamura, M., Colella, S., Yonekawa, Y., Kleihues, P., Ohgaki, H.: Loss of heterozygosity on chromosome 10 is more extensive in primary (de novo) than in secondary glioblastomas. Lab Invest. 2000; 80: 65-72. [ Links ]
28. Glick, R.P., Lichtor, T., Unterman, T.G.: Insulin-like growth factors in central nervous system tumors. J Neurooncol. 1997; 35: 315-325. [ Links ]
29. González-Gómez, P., Bello, M.J., Alonso, M.E., Arjona, D., Lomas, J., de Campos, J.M., Isla, A., Rey, J.A.: CpG island methylation status and mutation analysis of the RB1 gene essential promoter region and protein-binding pocket domain in nervous system tumours. Br J Cancer. 2003; 88: 109-114. [ Links ]
30. González-Gómez, P., Bello, M.J., Arjona, D., Lomas, J., Alonso, M.E., De Campos, J.M., Vaquero, J., Isla, A., Gutiérrez, M., Rey, J.A.: Promoter hypermethylation of multiple genes in astrocytic gliomas. Int J Oncol. 2003; 22: 601-608. [ Links ]
31. Halatsch, M.E., Schmidt, U., Behnke-Mursch, J., Unterberg, A., Wirtz, C,R.: Epidermal growth factor receptor inhibition for the treatment of glioblastoma multiforme and other malignant brain tumours. Cancer Treat Rev. 2006; 32: 74-89. [ Links ]
32. Ichimura, K., Schmidt, E.E., Goike, H.M., Collins, V.P.: Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene. 1996; 13: 1065-1072. [ Links ]
33. Joensuu, H., Puputti, M., Sihto, H., Tynninen, O., Nupponen, N.N.: Amplification of genes encoding KIT, PDGFRalpha and VEGFR2 receptor tyrosine kinases is frequent in glioblastoma multiforme. J Pathol. 2005; 207: 224-231. [ Links ]
34. Kaur, B., Tan, C., Brat, D.J., Post, D.E., Van Meir, E.G.: Genetic and hypoxic regulation of angiogenesis in gliomas. J Neurooncol. 2004; 70: 229-243. [ Links ]
35. Kleihues, P., Louis, D.N., Scheithauer, B.W., Rorke, L.B., Reifenberger, G., Burger, P.C., Cavenee, W.K.: The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol. 2002; 61: 215-225. [ Links ]
36. Knobbe, C.B., Reifenberger, G.: Genetic alterations and aberrant expression of genes related to the phosphatidylinositol-3'-kinase/protein kinase B (Akt) signal transduction pathway in glioblastomas. Brain Pathol. 2003; 13: 507-518. [ Links ]
37. Koichi Ichimura., Bondesson Bolin, M., Goike, H.M., Schmidt, E.E., Moshref, A., Collins, V.P.: Deregulation of the p14ARF/MDM2/p53 Pathway is a Prerequisite for Human Astrocytic Gliomas with G1-S Transition Control Gene Abnormalities. Cancer research. 2000; 60: 417-424. [ Links ]
38. Lamszus, K., Heese, O., Westphal, M.: Angiogenesis-related growth factors in brain tumors. Cancer Treat Res. 2004; 117: 169-190. [ Links ]
39. Laterra, J., Rosen, E., Nam, M., Ranganathan, S., Fielding, K., Johnston, P.: Scatter factor/hepatocyte growth factor expression enhances human glioblastoma tumorigenicity and growth. Biochem Biophys Res Commun.1997; 235: 743-747. [ Links ]
40. Layfield, L.J., Willmore, C., Tripp, S., Jones, C., Jensen, R.L.: Epidermal growth factor receptor gene amplification and protein expression in glioblastoma multiforme: prognostic significance and relationship to other prognostic factors. Appl Immunohistochem Mol Morphol. 2006; 14: 91-96. [ Links ]
41. López-Gines, C., Cerda-Nicolas, M., Gil-Benso, R., Pellin, A., López-Guerrero, A., Callaghan, R., Benito, R., Roldán, P., Piquer, J., Llacer, J., Barberá, J.: Association of chromosome 7, chromosome 10 and EGFR gene amplification in glioblastoma multiforme. Clin Neuropathol. 2005; 24: 209-218. [ Links ]
42. Machein, M.R., Plate, K.H.: VEGF in brain tumors. J Neurooncol. 2000; 50: 109-120. [ Links ]
43. Mollenhauer, J., Wiemann, S., Scheurlen, W., Korn, B., Hayashi, Y., Wilgenbus, K., von Deimling, A., Poustka, A.: DMBT1, a new member of the SRCR superfamily, on chromosome 10q25.3-26.1 is deleted in malignant brain tumours. Nat Genet. 1997; 17: 32-39. [ Links ]
44. Moriyama, T., Kataoka, H., Koono, M., Wakisaka, S.: Expression of hepatocyte growth factor/scatter factor and its receptor c-Met in brain tumors: evidence for a role in progression of astrocytic tumors. Int J Mol Med.1999; 3: 531-536. [ Links ]
45. Morrison, R.S., Yamaguchi, F., Saya, H., Bruner, J.M., Yahanda, A.M., Donehower, L.A., Berger, M.: Basic fibroblast growth factor and fibroblast growth factor receptor I are implicated in the growth of human astrocytomas. Neurooncol 1994; 18: 207-216. [ Links ]
46. Mueller, W., Mollenhauer, J., Stockhammer, F., Poustka, A., von Deimling, A.: Rare mutations of the DMBT1 gene in human astrocytic gliomas. Oncogene. 2002; 21: 5956-5959. [ Links ]
47. Nakamura, M., Watanabe, T., Klangby, U., Asker, C., Wiman, K., Yonekawa, Y., Kleihues, P., Ohgaki, H.: p14ARF deletion and methylation in genetic pathways to glioblastomas. Brain Pathol. 2001; 11: 159-168. [ Links ]
48. Nakamura, M., Yonekawa, Y., Kleihues, P., Ohgaki, H.: Promoter hypermethylation of the RB1 gene in glioblastomas. Lab Invest. 2001; 81: 77-82. [ Links ]
49. Ohgaki, H.: Genetic pathways to glioblastomas. Neuropathology. 2005; 25: 1-7. [ Links ]
50. Ohgaki, H., Dessen, P., Jourde, B., et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res. 2004; 64: 6892-6899. [ Links ]
51. Pérez-Ortiz, L., Galarraga, J., Gómez-Suárez, H., Tamayo-Suárez, J.D.: Classification of the astrocytic gliomas. Rev Neurol. 2000; 31: 1180-1183. [ Links ]
52. Reifenberger, G., Liu, L., Ichimura, K., Schmidt, E.E., Collins, V.P.: Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res. 1993; 53: 2736-2739. [ Links ]
53. Schmidt, M.C., Antweiler, S., Urban, N., Mueller, W., Kuklik, A., Meyer-Puttlitz, B., Wiestler, O.D., Louis, D.N., Fimmers, R., von Deimling, A.: Impact of genotype and morphology on the prognosis of glioblastoma. J Neuropathol Exp Neurol. 2002; 61: 321-328. [ Links ]
54. Sherr, C.J., Roberts, J.M.: CDK inhibitors: positive and negative regulators of G1-phase progression.Genes Dev. 1999; 13: 1501-1512. [ Links ]
55. Tohma, Y., Gratas, C., Biernat, W., Peraud, A., Fukuda, M., Yonekawa, Y., Kleihues, P., Ohgaki, H.: PTEN (MMAC1) mutations are frequent in primary glioblastomas (de novo) but not in secondary glioblastomas. J Neuropathol Exp Neurol. 1998; 57: 684-689. [ Links ]
56. Varela, M., Ranuncolo, S.M., Morand, A., Lastiri, J., De Kier Joffe, E.B., Puricelli, L.I, Pallotta, M.G.: EGF-R and PDGF-R, but not bcl-2, overexpression predict overall survival in patients with low grade astrocytomas. J Surg Oncol. 2004; 86: 34-40. [ Links ]
57. Visted, T., Enger, P.O., Lund-Johansen, M., Bjerkvig, R.: Mechanisms of tumor cell invasion and angiogenesis in the central nervous system. Front Biosci. 2003; 8: 289-304. [ Links ]
58. Ware, M.L., Berger, M.S., Binder, D.K.: Molecular biology of glioma tumorigenesis. Histol Histopathol. 2003; 18: 207-216. [ Links ]
59. Watanabe, K., Sato, K., Biernat, W., Tachibana, O., von Ammon, K., Ogata, N., Yonekawa, Y., Kleihues, P., Ohgaki, H.: Incidence and timing of p53 mutations during astrocytoma progression in patients with multiple biopsies. Clin Cancer Res. 1997; 3: 523-530. [ Links ]
60. Watanabe, T., Katayama, Y., Yoshino, A., Komine, C., Yokoyama, T.: Deregulation of the TP53/p14ARF tumor suppressor pathway in low-grade diffuse astrocytomas and its influence on clinical course. Clin Cancer Res. 2003; 9: 4884-4890. [ Links ]
61. Wong, A.J., Bigner, S.H., Bigner, D.D., Kinzler, K.W., Hamilton, S.R., Vogelstein, B.: Increased expression of the epidermal growth factor receptor gene in malignant gliomas is invariably associated with gene amplification. Proc Natl Acad Sci U S A. 1987; 84: 6899-6903. [ Links ]
62. Zumkeller, W., Westphal M.: The IGF/IGFBP system in CNS malignancy. Mol Pathol. 2001; 54: 227-229. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Carmen Franco / Juan A. Rey.
Laboratorio de Oncogenética Molecular.
Fundación para la investigación Biomédica.
Hospital Universitario La Paz.
Paseo de la Castellana 261.
28046 Madrid.
Recibido: 29-11-06.
Aceptado: 20-06-07
Abreviaturas. ADN: ácido desoxiribonucleico. CDK4: quinasa 4 dependiente de ciclina. DMBT1: Deleted in Malignant Brain Tumors. EGF: factor del crecimiento epidérmico. EGFR: receptor del factor del crecimiento epidérmico. EGFR vIII: variante 3 del receptor del factor del crecimiento epidérmico. E2F: factor de transcripción E2F. FGF: factor del crecimiento fibroblástico. FGFR: receptor del factor de crecimiento fibroblástico. FT: factor de transcripción. GBM: glioblastoma. GBM1: glioblastoma primario. GBM2: glioblastoma secundario. GST: gen supresor de tumores. HGF: factor de crecimiento hepatocítico. IGF: factor de crecimiento insulínico. LOH: pérdida de heterozigosidad. MDM2: proteína de unión a p53 MDM2. MGMT: metiltransferasa 6-O metilguanina. PDGF: factor del crecimiento plaquetario. PIP3: fosfatidil inositol 3 fosfato. PTEN: homólogo de la tensina y fosfatasa. P14arf: inhibidor la la kinasa 2B dependiente de ciclina. P16ink4: inhibidor la la kinasa 2A dependiente de ciclina. RBI: gen del retinoblastoma. SNC: sistema nervioso central. STAT: proteínas trasnductoras de señales y activadoras de la transcripción. TP53: proteína tumoral p53. TP73: proteína tumoral p73. VEGF: factor del crecimiento endotelial. VEGFR: receptor del factor del crecimiento endotelial

