










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkNeurocirugía
versión impresa ISSN 1130-1473
Neurocirugía vol.19 no.6 dic. 2008
Tumores de tronco cerebral en la población pediátrica
Brain-stem tumors in children
F.J. Villarejo; M.P. Ferrara; J.M. Belinchón; L. Madero*; B. Rivero; F. Cordobés; A. Pascual; C. Pérez-Díaz e I. de Prada**
Servicios de Neurocirugía, Oncología* y Anatomía Patológica**. Hospital Infantil Universitario Niño Jesús. Madrid.
Dirección para correspondencia
RESUMEN
Presentamos una serie de tumores de tronco cerebral diagnosticados y tratados en nuestro centro durante un período de 18 años. Se han revisado un total de 42 pacientes entre los años 1982 y 2006; 36 de ellos fueron intervenidos, realizándose una resección parcial en la mayoría de los casos.
Los tumores de tronco cerebral suponen una rara entidad patológica con muy mal pronóstico. En la mayoría de los casos no resulta posible una resección completa de la lesión, por lo que el principal objetivo de la cirugía es la reducción de masa tumoral y la descompresión. El mejor pronóstico se obtiene en los tumores de bajo grado de malignidad con mínimo déficit neurológico. La mayoría de estos tumores provocan la muerte en un breve período, habitualmente un año o menos desde el diagnóstico.
Palabras clave: Tumores de tronco cerebral. Clasificación de los tumores de tronco. Tumores pediátricos. Hidrocefalia. Resonancia magnética.
SUMMARY
We report a large series of brain-stem tumors seen during 18 years of at our hospital. We diagnosed and treated a total of 42 patients between 1988 and 2006; 36 of them were operated with partial resection in most cases.
Brain-stem tumors constitute a rare condition with very bad prognosis. A surgical complete resection of the mass is not possible in most cases, so the principal surgical objetive is reduction and descompression. The best prognosis is seen in patients with low grade tumors with minimal neurologic deficit. Most of these tumors cause death in a short period, usually one year or less.
Key words: Brain-stem tumors. Classification of brainstem neoplasms. Pediatric tumors. Hydrocephalus. MRI.
Introducción
El término tumor de tronco cerebral no describe a un solo tipo de tumor, sino a una variedad de neoplasias de origen glial que aparecen en el tronco encefálico, así como algunas lesiones benignas del tipo de las malformaciones vasculares.
Los tumores del tronco cerebral suponen un 10-25% de los tumores del sistema nervioso en el niño, y un 20 al 25% de los tumores infratentoriales. La media de edad al diagnóstico fue de 7 a 9 años, sin clara predilección por ningún sexo. En la mayoría de los casos, la historia natural de la enfermedad conlleva un desenlace fatal, con una mediana de sobrevida de 4 a 15 meses. Una supervivencia a los 5 años fue observada en un 30% de los pacientes1-3.
Anteriormente, los tumores de tronco suponían una entidad considerada de difícil tratamiento y de pronóstico fatal en la mayoría de casos. A lo largo de esta última década este concepto ha evolucionado como resultado de los grandes avances en técnicas de imagen, con la microcirugía y con la comprensión de su comportamiento biológico5.
En 1985, Epstein formuló una clasificación basada en el patrón de crecimiento observado en los estudios de resonancia magnética (RMN), que ha sido la empleada en este estudio para clasificar los resultados obtenidos. Dicha clasificación divide las lesiones en focales, difusas y cervicobulbares. Se consideran focales, las lesiones circunscriptas de menos de 2 cm. de diámetro sin edema asociado. Un tumor difuso es aquél con un gran componente hipodenso con o sin lesión focal. Los tumores cervicobulbares comprometen los dos tercios inferiores del bulbo y los segmentos rostrales de la médula espinal7. Poco más tarde, Hoffman describió la categoría de tumores dorsales exofíticos, los cuales tenían su origen dorsalmente en el bulbo, pero que presentaban un crecimiento exofítico hacia el cuarto ventrículo12. Barkovich, posteriormente, utilizó la clasificación de Epstein para describir una nueva manera de categorizar los tumores según los hallazgos de RMN3.
El presente trabajo analiza retrospectivamente una serie de 42 casos de tumores de tronco tratados en nuestro centro, incluyendo su diagnóstico anatomopatológico, clasificación y evolución posterior.
Material y métodos
Para el estudio se revisaron 42 casos atendidos en el servicio de Neurocirugía del Hospital Niño Jesús entre los años 1988 y 2006, de los cuales 20 fueron niños y 22 niñas. La media de edad de los pacientes fue 6,36 años, con un rango entre los 3 meses y los 12 años. Todos los pacientes, excepto seis, fueron intervenidos quirúrgicamente obteniendo así un diagnóstico anatomopatológico posterior.
Resultados
Signos y sintomatología
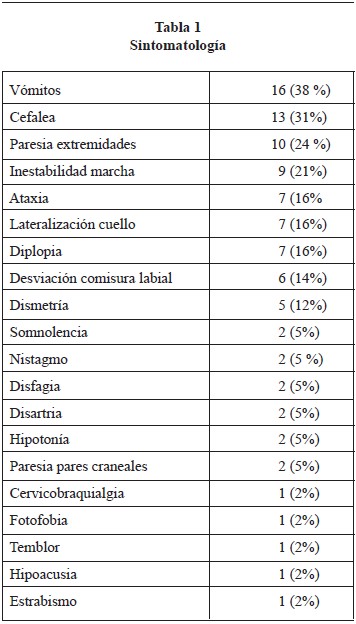
La sintomatología de inicio más frecuente encontrada fueron los vómitos, cefalea, paresia de extremidades y alteraciones de la marcha. Los signos más frecuentes fueron cerebelosos y de afectación de pares craneales (Tabla 1). Debe mencionarse que el 98% de los casos presentaban un cuadro de hidrocefalia asociada.
Localización de las lesiones
La localización más frecuente, según el estudio por RMN, fue en el bulbo con 19 casos (45%), seguido de la protuberancia con 12 casos (30%) y del mesencéfalo con 11 casos (25%).
Utilizando la clasificación de Epstein los resultados fueron: 20 lesiones difusas, 13 focales, 7 dorsales exofíticas y ninguno cervicobulbar (Tabla 2).

Anatomía patológica
Los resultados anatomopatológicos predominantes fueron: astrocitoma grado I 11 casos, grado II 4 casos, grado III 4 casos y grado IV 16 casos y además, un caso de malformación vascular de protuberancia. El resto de los casos corresponde a pacientes no intervenidos ni biopsiados, salvo dos casos en los que se perdió el seguimiento por realizar interconsulta con otro centro.
Evolución
Para conocer la evolución de los pacientes a largo plazo fue necesario contactar con los familiares o con el paciente. De todos los casos estudiados, los pacientes con diagnóstico de astrocitoma grado I (11 casos) continúan vivos entre 1 y 17 años después de la intervención. Los pacientes con gliomas de grado II o superior no sobreviven en ningún caso; la media de supervivencia fue de 9,8 meses desde el diagnóstico.
Discusión
El gran cambio en la última década con respecto a este tema ha sido que el pronóstico de los pacientes portadores de tumores de tronco cerebral depende de su localización y de su patrón de crecimiento9. El gran aporte de la RMN fue poder diferenciar las lesiones difusas del resto de los tumores. Debido al comportamiento maligno e invasivo de estas lesiones el tratamiento quirúrgico es rara vez beneficioso y por el contrario parece altamente peligroso. De ahí que tradicionalmente han sido tratados con radioterapia convencional fraccionada y quimioterapia adyuvante con relativamente escaso éxito terapéutico7. En los últimos años, sin embargo, se ha visto que ciertos grupos de tumores de tronco responden considerablemente tras un tratamiento quirúrgico invasivo.
En la serie analizada por Epstein en 19867 y luego revisada en 19936, todos los pacientes con tumores difusos fueron gliomas de alto grado. Ninguno de ellos se vio beneficiado por la cirugía y todos fallecieron entre los 6 a 12 meses posteriores a la misma. En cuanto a los pacientes con lesiones focales, la gran mayoría correspondieron a lesiones de bajo grado. Todos los pacientes con tumores de la unión cervicobulbar tuvieron astrocitomas grado I y II, sin presentar mortalidad operatoria alguna. Con lo cual queda demostrado que los tumores que comprometen la región cervicobulbar son generalmente benignos, y suelen desplazar más que invadir las estructuras adyacentes. Con tratamiento quirúrgico agresivo estos pacientes pueden prolongar su sobrevida. Con respecto a las lesiones dorsales exofíticas, también se verían beneficiadas por la cirugía. Lo mismo sucedería con ciertas lesiones focales ubicadas más rostralmente. Es poco factible en todos estos casos lograr una exéresis completa, ya que estas lesiones se originan intrínsecamente en el parénquima del tronco cerebral. Es muchas veces este tumor residual tema de controversia, en cuanto a si aplicar o no terapia radiante, o si reservar la misma para casos con progresión tumoral6,9.
Ante la decisión terapéutica es imprescindible evaluar en conjunto la duración de la enfermedad, la evolución de la misma, la severidad de los síntomas y los resultados de los estudios con neuroimagen. Todos los tumores difusos presentan sintomatología precoz, típica y florida, como disfunción de los pares craneales, generalmente múltiple y bilateral, y signos de vías largas, y su evolución generalmente es rápidamente progresiva. El edema difuso se observa de manera constante en la RMN, lo cual sugiere que lo que se observa puede corresponder a infiltración tumoral propiamente dicha más que edema. Poseen su epicentro típicamente en la protuberancia, y no presentan un límite claro, se extienden rostral y caudalmente. La ausencia de un componente focal que capte contraste no implica que el tumor sea de bajo grado. Si se logra visualizar un anillo de parénquima rostral al tumor, podría ser considerado como una lesión focal. Los pacientes con tumores focales suelen debutar con mínimos síntomas y con un curso más insidioso de la enfermedad. Son más frecuentes a nivel bulbar. Aquellos que debutan con sintomatología típica y progresiva podrían ser tumores focales de alto grado. Los tumores de la unión cervicobulbar debutan con alteración de los pares craneales bajos asociado a hemi o tetraparesia y espasticidad5-7.
En cuanto al patrón de crecimiento tumoral, Epstein sostiene que existen barreras anatómicas tanto en la unión cervicobulbar como en la bulboprotuberancial, las cuales serían relativamente impermeables a la expansión de un tumor de bajo grado, pero esta barrera se vería seriamente alterada ante la presencia de un tumor de alto grado6.
Siempre la evolución clínica es el factor pronóstico aislado más importante si no existe correlación con la RMN. Si el cuadro clínico y la RMN son concluyentes, muchos autores coinciden en no realizar biopsia como requisito para comenzar tratamiento oncológico. Por el contrario consideran que la cirugía expone al paciente a un riesgo de morbilidad y mortalidad injustificado5,7. En un estudio multicéntrico doble ciego publicado recientemente, se compararon los resultados obtenidos en 142 pacientes con enfermedad del tronco cerebral, utilizando RMN versus biopsia. Concluyeron que la sensibilidad de la RMN en el diagnóstico de tumores de tronco es de 0.94 con un valor predictivo positivo de 0.96. La tasa de morbilidad asociada a la biopsia fue de 3.2% y una mortalidad entre el 0.05 y el 2.6%11.

Figura 1. Astrocitoma protuberancia.

Figura 2. Astrocitoma bulbo-medular.

Figuras 3 y 4. Astrocitoma protuberancia, IRM e imagen operatoria.
Conclusión
Actualmente, en la mayoría de los casos es posible realizar un diagnóstico específico antes de la cirugía. La clave está en operar aquellos pacientes con mínima disfunción neurológica y con lesiones de bajo grado. Ningún paciente con un tumor maligno se ha beneficiado significativamente con la intervención, ya que todos fallecen dentro de los primeros 12 meses7. Contrariamente, con un tratamiento quirúrgico agresivo, los pacientes portadores de tumores en la región cervicobulbar, lesiones focales y dorsales exofiticas pueden presentar largas supervivencias sin recidivas9.
Bibliografía
1. American Association of Neurological Surgeons, Pediatric Neurosurgery, 2nd edition, W.B. Saunders, USA, 1989 pp 357-365. [ Links ]
2. American Association of Neurological Surgeons, Pediatric Neurosurgery, 4th edition, W.B. Saunders, USA, 2001 pp 81-86. [ Links ]
3. Barkovich, A., Krischer, J., Kun, L., Packer, R., Zimmerman, R., Freeman, C., et al.: Brain Stem Gliomas: a classification system based on magnetic resonance imaging. Pediatr Neurosurg 1990-91; 16: 73-83. [ Links ]
4. Broniscer, A., Gallar, A.: Supratentorial high-grade astrocytoma and diffuse brainstem glioma: two challenges for the pediatric oncologist. The Oncologyst 2004; 9: pp 197-206. [ Links ]
5. Epstein, F., Constantini, S.: Practical decisions in the treatment of pediatric brain stem tumors. Pediatr Neurosurg 1996; 24: 24-34. [ Links ]
6. Epstein, F. and Farmer, J.: Brain-stem glioma growth patterns. J Neurosurg 1993; 78: 408-412. [ Links ]
7. Epstein, F., Mc Leary, E.: Intrinsic brain-stem tumors of childhood: surgical indications. J Neurosurg 1986; 64: 11-15. [ Links ]
8. Jallo, G.l., Roonprapunt, C., et al.: Current management of brainstem Gliomas. Annals of Neurosurgery, 2003; 3: pp 1-17. [ Links ]
9. Pollack, I., Hoffman, H., Humphreys, R., and Becker, L.: The long-term outcome after surgical treatment of dorsally exophytic brain-stem gliomas. J Neurosurg 1993; 78: 859-863. [ Links ]
10. Ragheb, J., Epstein, F.: The surgical classification and management of brainstem tumors in children. International Pediatrics vol. 15, 2000 pp 15-20 [ Links ]
11. Schumacher, M., Schulte-Monting, J., Stoeter, P., Warmuth-Metz, M., and Solymosi, L.: Magnetic resonance imaging compared with biopsy in the diagnosis of brainstem diseases of childhood: a multicenter review. J Neurosur (2 suppl pediatrics) 2007; 106: 111-119. [ Links ]
12. Stroink, A., Hoffman, H., Hendrick, E. and Humphreys, R.: Diagnosis and management of pediatric brain-stem gliomas. J Neurosurg 1986; 65: 745-750. [ Links ]
13. Winn, R.: Youmans Neurological Surgery, fifth edition. WB Saunders, USA 2004, pp 3663-3670. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
José María Belinchón de Diego.
Hospital General Universitario de Valencia.
Servicio de Neurocirugía.
AV. Tres Cruces sn.
46014 Valencia.
Recibido: 17-08-07.
Aceptado: 02-05-08

