










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
Desde 2017 la normativa europea sobre productos sanitarios (PS) es el Reglamento 2017/745 sobre PS1, en adelante “Reglamento”. La legislación suiza en productos sanitarios (PS) venía siendo equivalente a la europea desde 2002 a través de la Ordenanza de PS (Medical Device Ordinance, MedDO)2 junto con el Acto Federal de Medicamentos y Productos Sanitarios3. Esta equivalencia era posible gracias al Acuerdo de Reconocimiento Mutuo (ARM) de certificados de conformidad Unión Europea-Suiza4. Este ARM tiene como base legal las Directivas de PS5-7. Esta equivalencia se perdió al entrar en vigor el 26 de mayo de 2021 el Reglamento 745/2017 sobre PS1, en adelante “Reglamento, deroga la anterior Directiva 93/42 de PS6 y la Directiva 90/385 de Productos Sanitarios Implantables Activos (ARM)7.
En consecuencia, y como medida de contingencia, el 19 de mayo el Consejo Federal Suizo aprobó una revisión completa del MedDO, así como provisiones suplementarias junto con una nueva normativa de ensayos clínicos con PS8.
Ambas partes, Suiza y la Unión Europea (UE), han elaborado un nuevo ARM para PS que está finalizado y listo para firmarse, pero la UE vincula su firma al avance en la negociación del Acuerdo Marco Institucional9.
En lo que respecta a los Productos Sanitarios de Diagnóstico in Vitro (PSDIV), durante la vigencia de la Directiva europea5 no será necesaria legislación adicional en Suiza hasta que el nuevo Reglamento de PSDIV entre en vigor (RE 746/2017 sobre PSDIV)10el próximo 26 de mayo de 2022.
Por otro lado, está la situación con el Reino Unido (RU). El RU está formado por Gran Bretaña (Escocia, Inglaterra y Gales) e Irlanda del Norte (IN). A diferencia de Suiza, el RU sí que tenía condición de Estado miembro, ya que formaba parte de la UE desde el 1 de enero de 197311.
A raíz del referéndum celebrado el 23 de junio de 2016 en el que se decidió el abandono de la UE, el gobierno invocó el artículo 50 del Tratado de Funcionamiento de la Unión Europea notificando a la Comisión Europea (CE) su intención de abandonar la misma el 29 de marzo de 2017, iniciándose así el proceso conocido como Brexit (Britain exit). El 24 de diciembre de 2020 la UE y RU firmarían un Acuerdo de Comercio y Cooperación que definiría las futuras relaciones. Este acuerdo entró en vigor el 1 de mayo de 202112. Hasta ese momento, el RU se había regido por la normativa europea en PS, no llegándose a implementar los nuevos Reglamentos al entrar en vigor tras el periodo de transición.
Un punto clave del Acuerdo de Retirada de RU de la UE fue el Protocolo de Irlanda del Norte13. Este protocolo se implementó para evitar la creación de una frontera física que separara IN del resto de Irlanda y se preservara la integridad del Mercado Único de mercancías de la UE14. Además, por medio de este protocolo ciertos productos, incluyendo los PS, deben cumplir con la legislación de la UE y el RU más allá del periodo transitorio15.
El objetivo de este artículo es comparar las medidas que se han tomado por parte de la UE, Suiza y RU para mantener la continuidad de mercado cumpliendo con los requisitos regulatorios del Reglamento.
Métodos
Para realizar este trabajo se han revisado las webs oficiales de la CE, la Agencia Española del Medicamento y Productos Sanitarios (AEMPS), la Regulatory Agency for Medicines and Health Products (Swissmedic) y la Medicines and Healthcare Products Regulatory Agency (MHRA). Además, se ha realizado una búsqueda en PubMed sobre los ARM en PS en Suiza y RU países que no ha reportado resultados. También se han realizado búsquedas en internet (Google), tanto en inglés como en castellano, utilizando palabras clave como “Mutual Recognition Agreement medical devices Switzerland”, “Mutual Recognition Agreement medical devices UK”,“Northern Ireland Protocol medical devices” para el intervalo temporal desde enero de 2021 hasta diciembre de 2021. Se ha considerado la información procedente de páginas web de Organismos de Evaluación de la Conformidad (ON) y agencias reguladoras.
Resultados
La información obtenida se ha presentado en función de los países y en epígrafes para facilitar su comparación.
Unión Europea-Suiza
La UE considera a Suiza como tercer país en sus relaciones comerciales con PS al cesar la vigencia del ARM4 el 26 de mayo de 2021. El marco normativo de Suiza actualmente se basa en la MedDO revisada y las medidas suplementarias aprobadas el 19 de mayo de 2021 por el Consejo Federal Suizo junto con una nueva normativa sobre ensayos clínicos con PS (CTO MedD)8. Ambas modificaciones entraron en vigor el 26 de mayo de 20212. Por otro lado, en virtud del art. 82.3 del Acto Federal de Medicamentos y PS, el Consejo Federal suizo puede implementar medidas de la CE con respecto al Reglamento1 aplicables en Suiza siempre que sean directamente aplicables los actos delegados y de ejecución de la CE16.
Requisitos para los fabricantes y representantes autorizados
Desde el 26 de mayo de 2021, los fabricantes europeos que deseen poner en el mercado o poner en servicio un PS en Suiza deben hacerlo a través de un importador, además de nombrar a un representante autorizado suizo (CH-REP). El CH-REP se requiere para todos los PS, incluyendo los PS a medida, los kits y sistemas, así como los que no tienen un propósito médico según el anexo 1 del MedDO.
Los operadores económicos (fabricantes, importadores y CH-REP) deben registrarse en los 3 meses siguientes a introducir un producto en el mercado en Swissmedic. Tras el registro se les asignará un número de identificación único (swiss single registration number, CHRN)17, el equivalente del SRN europeo (European single registration number). Existen los siguientes periodos de gracia para que el fabricante designe un CH-REP9:
Para dispositivos de clase III, dispositivos implantables de clase llb y dispositivos implantables activos: hasta el 31 de diciembre de 2021.
Para los dispositivos no implantables de clase llb y los PS clase lla: hasta el 31 de marzo de 2022.
Para los dispositivos de clase I: hasta el 31 de julio de 2022.
Para los kits y sistemas: hasta el 31 de julio de 2022.
Estos plazos ampliados se aplican también a Islandia y Noruega, pero no a Liechtenstein debido al tratado aduanero entre Suiza y este país18. Estos plazos tampoco se aplican a los importadores9.
La tabla 1 resume las principales diferencias entre los diversos tipos de certificación.
Tabla 1. Características de los diferentes procedimientos de evaluación de productos sanitarios
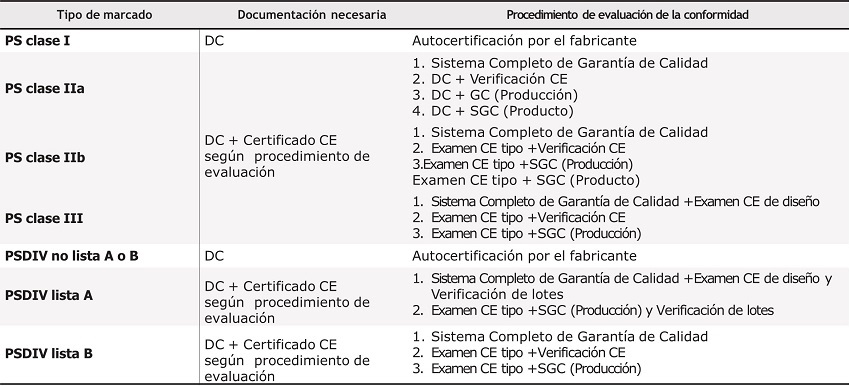
DC: declaración de conformidad; PS: producto sanitario; PSDIV: Productos Sanitarios de Diagnóstico in Vitro; SGC: sistema garantía de calidad.
De forma equivalente, los fabricantes suizos o los fabricantes de terceros países con un CH-REP tienen que designar a un representante autorizado en la UE (EC-REP)19. Esta nueva información se tiene que actualizar en la declaración UE de conformidad20. Además, desde el 26 de mayo de 2021 los distribuidores necesitan licencia de importación para ejecutar estas transacciones comerciales.
Registro de PS
En Suiza es necesario hacer una comunicación a Swissmedic desde julio de 2020 por parte de los fabricantes de los PS a medida, PS reacondicionados o reetiquetados, PS de clase I y aquellos de clase I que cambian de clasificación con el Reglamento21.
En la UE, el registro de los PS se ha venido haciendo a nivel nacional en las respectivas bases de datos de cada Estado miembro. El registro en la base de datos europea European Database on Medical Devices (EUDAMED3)22 todavía no se ha completado. EUDAMED es la futura base de datos europea para PS. Con ella se mejorará la transparencia y la coordinación de la información sobre PS. Está formada por seis módulos (registro de los agentes económicos, registro UDI/PS, Organismos Notificados y Certificados, Investigaciones Clínicas y estudios de funcionamiento, Vigilancia y Vigilancia Post-Autorización, Vigilancia de Mercado). Esta base de datos almacena la información necesaria para el control de mercado y tener un sistema de vigilancia eficiente a nivel europeo. Actualmente, en España la notificación debe realizarse a Comunicaciones de Comercialización de Productos Sanitarios (CCPS).
Etiquetado y marcado de conformidad
En Suiza, el etiquetado y las instrucciones de uso deben estar escritas en las tres lenguas oficiales (alemán, francés e italiano)17. Debe aparecer la marca de conformidad suiza (“MD”) en el etiquetado, aunque también se permite el marcado CE17,23.
El CH-REP debe figurar en el cartonaje exterior junto con el símbolo “CH-REP”, pero no es obligatorio que aparezca en el producto, en las instrucciones de uso o en los documentos que acompañan al producto, de acuerdo con una nota informativa de Swissmedic24.
En la UE, la nota emitida por la CE dirigida a las partes interesadas el 26 de mayo de 202119 recordaba la obligatoriedad del registro y normas de etiquetado vigentes para los PS suizos.
En el caso de España, la AEMPS estableció el 30 de septiembre de 2021 como fecha límite para actualizar el etiquetado e instrucciones de uso con el nuevo EC-REP u ON mediante una nota informativa (Nota PS 20/21)20. Esta fecha se ha retrasado al 30 de septiembre de 2022 (Nota PS 29/21)25.
Organismos de evaluación de la conformidad y certificados
Los certificados emitidos antes del 25 de mayo de 2017 serán válidos hasta su caducidad, no más allá del 26 de mayo de 2022; y los certificados emitidos bajo la anterior legislación desde el 25 de mayo de 2017 se mantendrán válidos hasta su caducidad, no más allá del 26 de mayo de 202417.
Los certificados CE de ON suizos perdieron su validez el 26 de mayo de 202119, ya que la UE no los reconoce. En las declaraciones de conformidad deben actualizarse los datos del EC-REP y número de certificado.
Productos Sanitarios “legacy”
Los PS conocidos como legacy devices son aquellos PS puestos en el mercado legalmente cumpliendo con las disposiciones de las Directivas6,7antes de la entrada en vigor del Reglamento y que se les permite seguir comercializándose. Esta situación particular está incluida en el artículo 120 del Reglamento6,7. Éstos son:
-
- Dispositivos de clase I con una declaración de conformidad emitida antes del 26 de mayo de 2021 que necesitan un certificado según la nueva normativa (por ejemplo, instrumental reutilizable, o PS que pasan a tener una clase mayor con el Reglamento).
- Dispositivos con un certificado CE válido bajo la anterior normativa.
Estos productos pueden permanecer en el mercado suizo tras el 26 de mayo de 2021 hasta que el certificado expire, pero no más tarde del 26 de mayo de 2024. Aunque pueden permanecer en la cadena de distribución hasta el 26 de mayo de 2025. Deben cumplir con los requisitos de la directiva de PS o ARM, siempre que no hayan sufrido cambios en su diseño ni propósito26.
En el caso europeo, se entenderá por productos sanitarios legacy devices los PS cuyo fabricante o representante autorizado es suizo, o que tienen un certificado de conformidad emitido por un ON suizo. Con la publicación de la Nota a las partes interesadas de la Comisión el pasado 26 de mayo de 202119, esta clase de dispositivos no pueden continuar en el mercado europeo
Unión Europea-Reino Unido
El RU está formado por Gran Bretaña e IN. Se analiza la situación de cada región por separado.
Situación en Gran Bretaña
Actualmente las disposiciones reguladoras inglesas se basan en las tres antiguas directivas de PS5-7 traspuesta en la regulación inglesa de PS de 2002 (UK MDR 2002, por sus siglas en inglés)27.
-
- Requisitos para los fabricantes y representantes autorizados
Los fabricantes que deseen poner en el mercado inglés un PS tienen que registrarse en la MHRA del RU y designar a una persona responsable en RU (UKRP, por sus siglas en inglés). No es necesario un UKRP para importadores y distribuidores, pero pueden actuar como tal. Cuando el importador no sea el UKRP, éste debe informar al UKRP de su intención de importar PS, y el UKRP debe notificar a la MHRA sus importadores de PS27
En la UE, los fabricantes ingleses tienen las mismas exigencias que los suizos o con CH-REP.
-
- Registro de PS
Todos los PS, PSDIV y PS a medida deben registrarse en la MHRA antes de ponerse en el mercado. Si un PS ya estaba registrado en la MHRA, no hay que volver a registrarlo (fabricantes de IN27). Existen periodos de gracia para realizar el registro27:
Los PS IA, PS clase III, PS clase IIb implantables y los PSDIV de la lista A deben registrarse a partir del 1 de mayo de 2021.
Los PS clase IIb no implantables, los PS clase IIa, los PSDIV lista B y los de autodiagnóstico deben registrarse a partir del 1 de septiembre de 2021.
Los dispositivos de clase I, los PSDIV generales (que actualmente no necesitan estar registrados) deben registrarse a partir del 1 de enero de 2022.
Los fabricantes de PS clase I, PS a medida y PSDIV general que antes del 1 de enero de 2021 necesitaban registrarse deben seguir haciéndolo.
En la UE, se requiere lo mismo que a los fabricantes suizos o con CH-REP.
-
- Etiquetado y marcado de conformidad
Deben llevar el marcado UKCA (UK Conformity Assessed) o CE, según la legislación bajo la que estén certificados, y el número del ON u Órgano de Evaluación de la Conformidad inglés (UK Conformity Assessment Body, UKCAB) cuando aplique27.
Los PS clase I, PSDIV generales pueden autocertificarse con el marcado UKCA, que hasta el 30 de junio de 2023 es voluntario y posteriormente será obligatorio27.
Se permite que ambos marcados (UKCA y CE) estén presentes, incluso después de esa fecha, pero el nombre y dirección de la UKRP tiene que aparecer junto con el marcado UKCA (incluso los PS con marcado dual)27. Por otro lado, el marcado UKCA no está reconocido en la UE o IN, donde se requiere el marcado CE (o UKNI-CE en IN)27. En España, la AEMPS inicialmente se concedió un plazo hasta el 30 de junio de 2020 para la adecuación del etiquetado e instrucciones de uso28 que se retrasó hasta el 25 de mayo de 2021 ante la dificultad de su implementación29.
-
-Organismos de evaluación de la conformidad y certificados
La MHRA puede designar UKCAB para hacer evaluaciones de conformidad para Gran Bretaña en base al UK MDR 2002, pero no con relación al marcado CE, a excepción del marcado UKNI-CE para IN, que sólo es válido en esa región27. La MHRA convirtió automáticamente los antiguos ON en UKCAB27.
Además, el gobierno ha creado una nueva base de datos llamada UK Market Conformity Assessment Bodies para reemplazar al sistema de información europeo NANDO27.
En referencia a los certificados CE emitidos bajo un ON reconocido por la UE (ON incluidos en NANDO), serán válidos hasta el 30 de junio de 202327.
En la UE no se reconocen a los ON ingleses desde el 26 de mayo de 2021.
Irlanda del Norte
Por el Protocolo de IN, las exigencias para comercializar un dispositivo aquí son diferentes de las de Gran Bretaña. Debido a su situación geográfica particular es de aplicación tanto la normativa inglesa como europea.
-
– Requisitos para los fabricantes y representantes autorizados
Los fabricantes y representantes autorizados ubicados en la UE/Espacio Económico Europeo deben designar a un único UKRP para los dispositivos que se comercialicen en IN. No es necesario si el fabricante tiene su sede en Gran Bretaña o en IN o si tiene un representante autorizado localizado en IN (INRA, por sus siglas en inglés). Tampoco en caso de comercializar PS clase I, a medida o PSDIV generales registrados por una autoridad competente de la UE.
Del mismo modo, los fabricantes de Gran Bretaña que quieran introducir un PS en el mercado de IN deben nombrar un EC-REP o un INRA15. Es posible que una sola entidad actúe como INRA y como UKRP.
En aquellos casos donde un importador de IN no es el INRA o UKRP, el importador tiene que informar a los correspondientes INRA o UKRP de su intención de importar PS, y éstos deben informar a la MHRA de la lista de importadores15.
En lo que respecta a la normativa europea, los fabricantes ubicados en IN no requieren un EC-REP. En el caso de que el dispositivo necesite una evaluación por parte de un ON, éste debe ser europeo15. Los fabricantes de RU o de terceros países podrán nombrar un EC-REP en IN para la UE.
-
– Registro de PS
La mayoría de los PS, a medida y PSDIV generales que se comercializan en IN tienen que registrarse en la MHRA. Esto va a depender de la localización del fabricante y del representante autorizado y de la clase del dispositivo y están sujetos a los mismos periodos de gracia que en RU15.
Cuando se designa un representante autorizado con sede en IN, éste debe registrar todas las clases de dispositivos en la MHRA. En cambio, si su sede está en la UE, el fabricante debe registrar todas las clases de PS que no sean clase I, a medida o PSDIV generales.
En lo relativo a la UE, no hay exigencias adicionales a las del Reglamento.
-
– Etiquetado y marcado de conformidad
El marcado UKCA no es válido en IN. En este territorio, para aquellos dispositivos que necesiten una evaluación por una tercera parte, además del marcado CE, llevarán el UKNI si dicha evaluación la ha hecho un UKCAB27. El marcado UKNI no puede estar solo. Si la evaluación la realiza un ON reconocido por la UE, únicamente llevará el marcado CE. Por tanto, el marcado UKNI aparecerá en un dispositivo cuando éste vaya a comercializarse en IN, necesite una evaluación de conformidad y ésta la haya realizado un UKCAB27.
En lo concerniente a la UE, no se acepta el macado UKNI, y los PS destinados al mercado de la UE llevarán solo el marcado CE27.
-
– Organismos de evaluación de la conformidad y certificados
Los ON del RU pueden realizar evaluaciones de conformidad únicamente para el mercado de IN pero no para la UE. Para poder poner el marcado CE válido en un dispositivo para IN y la UE éste debe haber sido evaluado por un ON reconocido en la UE.
El gobierno inglés ha garantizado la comercialización de los PS de IN en el mercado de RU. Esto significa que los PS con el símbolo de conformidad de IN se consideran válidos para comercializar en todo el RU, incluso más allá del 30 de junio de 2023.
En lo referente a la normativa europea, los ON irlandeses certifican conforme al Reglamento.
La tabla 2 resume los cambios principales en la puesta en mercado en la UE, Suiza y RU tras la entrada en vigor del nuevo Reglamento.
Tabla 2. Resumen de los principales cambios y medidas tomadas por la Unión Europea Suiza y reino Unido
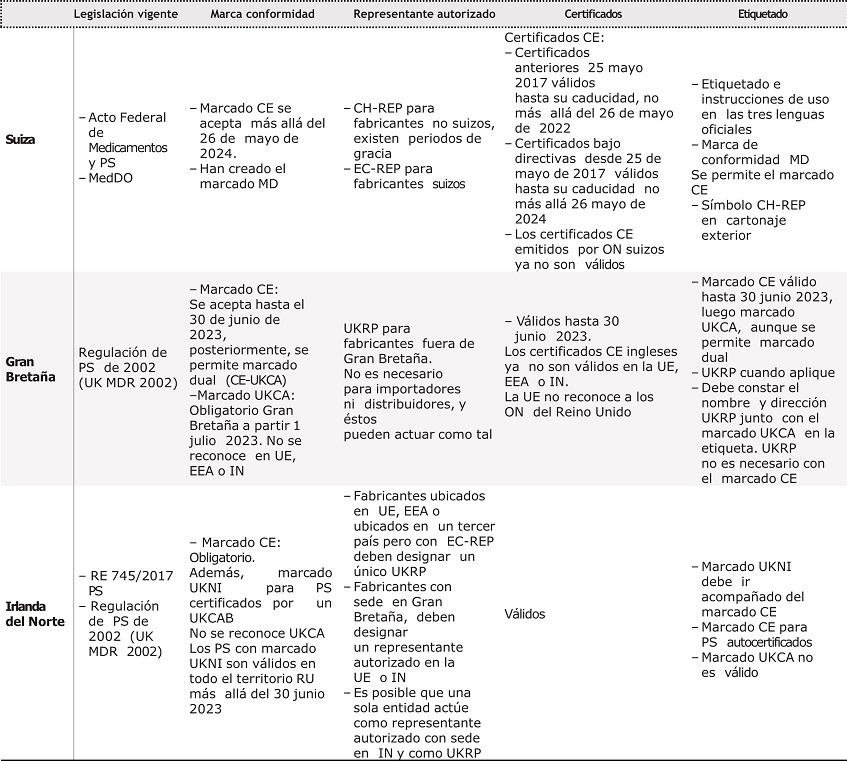
CH-REP: representante autorizado suizo; EC-REP: representante autorizado en la Unión Europea; EEA: Espacio Económico Europeo; IN: Irlanda del Norte; MedDO: Medical Device Ordinance; ON: Organismo de Evaluación de la Conformidad; PS: productos sanitarios; RU: Reino Unido; UE: Unión Europea; UK MDR 2002: Medical Device Regulation 2002; UKCA: Marca de Conformidad en Gran Bretaña; UKCAB: Órgano de Evaluación de la Conformidad inglés; UKRP: representante autorizado en el Reino Unido
Discusión
El cese del libre comercio de PS entre la UE, y Suiza y RU supone un perjuicio para los operadores económicos y los pacientes. Este artículo compara las medidas que se han tomado, en este momento de gran confusión, para cumplir con los nuevos requisitos exigidos, tras la entrada en vigor del Reglamento, en la puesta en el mercado de PS en UE, Suiza y RU.
Las dos principales consecuencias de convertirse Suiza y RU en terceros países son, por una parte, la posibilidad de desabastecimiento y, por otra, la obligada necesidad de creación de registros de operadores económicos y dispositivos y otras estructuras/bases de datos que permitan el control de su mercado interno en estos países.
La UE, por su tamaño, está en una posición ventajosa respecto a Suiza y RU. Ello le ha permitido cesar, de forma inmediata y unilateral, la validez de los certificados CE, emitidos por ON de estos países y, por tanto, hacer que los PS procedentes de estos países no estén legalmente en el mercado. Sin embargo, la situación inversa no ha sido posible.
La pérdida simultánea de validez de los certificados CE conlleva una carga de trabajo extra para el resto de los ON europeos y un retraso en la emisión de nuevos certificados. Las empresas que no lo hayan previsto con antelación, habrán visto paralizadas sus relaciones comerciales hasta la obtención de los correspondientes certificados y actualización de la documentación técnica
Por otro lado, según el Memorándum de Sidley Austin EU/EEA Market Access for “Swiss Legacy Devices” post abandonment of Swiss EU MRA 30 “la decisión unilateral de la Comisión de cesar la aplicación del ARM y la aparente retirada retrospectiva por parte de la Comisión del reconocimiento mutuo de los PS legacy expresada en la nota a las partes interesadas, son contrarias a la legislación de la UE, al ARM y al derecho de la Organización Mundial del Comercio”30. Aunque este punto de vista es válido, únicamente se podría considerar para los legacy devices con certificados CE de ON no suizos. Hasta el momento, no se ha encontrado información sobre la posición de la CE respecto a este planteamiento.
Por otro lado, está la necesidad de crear registros de los operadores económicos y de PS que les permitan realizar un control y vigilancia de mercado correcto. La creación de estas bases de datos debe ser prioritaria, así como negociar algún tipo de acceso a EUDAMED3, ya que tanto Suiza como RU están dentro del continente europeo y cualquier incidente ocurrido en él puede repercutir en estos países.
Es de esperar que tanto Suiza como RU se adaptarán a los nuevos reglamentos a través de su regulación interna, ya que cualquier otra decisión sería poner trabas adicionales al comercio, lo que finalmente repercutiría en la salud de los pacientes.
Por tanto, la importancia del presente trabajo es consolidar en un único documento los principales cambios regulatorios y periodos de transición concedidos a los operadores económicos y agencias reguladoras a raíz de la escisión normativa en el campo de los PS de Suiza y RU desde el 26 de mayo de 2021.
En conclusión, a pesar de que los motivos de la separación de la UE de RU y Suiza son diferentes, ambos países se han visto obligados a adaptarse a las disposiciones del Reglamento para evitar un más que previsible desabastecimiento.
La UE ha hecho valer su posición de fuerza anulando con carácter inmediato los certificados CE emitidos por estos países y exigiendo una nueva certificación por ON europeos reconocidos, aunque se han dado periodos de gracia. Además, en el caso de Suiza, existen dudas sobre la legalidad de esta medida.
Por el contrario, tanto Suiza como RU se han visto obligadas a establecer periodos transitorios más largos para que las agencias reguladoras y los operadores económicos pudieran adaptar los dispositivos a las nuevas legislaciones nacionales. Es decir, estos países siguen aceptando PS con marcado CE y certificados emitidos por ON europeos, en Suiza permanentemente y en RU hasta el 30 de junio de 2023.
Es de esperar que conforme se vaya avanzando en la implementación del Reglamento en la UE, estos países tomen medidas de contingencia adicionales. El presente trabajo pone de manifiesto que serán necesarios nuevos estudios para abordar las medidas finales tomadas.
Aportación a la literatura científica
Este trabajo clarifica el nuevo escenario regulatorio entre la Unión Europea, Suiza y Reino Unido en el ámbito de los productos sanitarios. El cese del Acuerdo de Reconocimiento Mutuo con Suiza ha coincidido temporalmente con la entrada en vigor del nuevo Reglamento 745/2017 de productos sanitarios1. Además, desde enero de 2021 el Reino Unido ya no pertenece a la Unión Europea. Los operadores económicos tienen que adecuarse a los nuevos requisitos del Reglamento y de cada país en particular, sin que éstos estén claramente detallados en un documento oficial.

