










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista de la Sociedad Española del Dolor
versión impresa ISSN 1134-8046
Rev. Soc. Esp. Dolor vol.11 no.5 Madrid jul. 2004
ORIGINAL
Relación del efecto analgésico de fentanilo agudo con la regulación
a la alta de los receptores 5-HT1A cerebrales en la rata
I. Bellido1, M. J. Castro2, A. Gómez-Luque1-3 y F. Sánchez de la Cuesta1
| Bellido I, Castro MJ, Gómez-Luque A, Sánchez de la Cuesta F. Relationship between the analgesic effect of acute fentanyl and upregulation of brain 5-HT1A receptors in the rat. Rev Soc Esp Dolor 2004; 11: 270-281.
| ||
| SUMMARY 5-HT1A agonists have analgesic effects. The analgesic effect of µ agonists can be blocked by selective 5-HT1A antagonists. In order to determine the mechanism that produces the synergies observed between µ and serotoninergic 5-HT1A receptors in terms of their antinociceptive effect, we determined the analgesic effect of fentanyl after ap-plying a painful thermal and mechanical stimulus in the rat, and related it with the affinity and the maximum density of 5-HT1A receptors in thirteen brain areas using autoradiographic techniques. Fentanyl showed a dose- and time-dependant analgesic effect with the two nociceptive stimuli. In addition to its analgesic effect, fentanyl caused an up-regulation of 5-HT1A receptors, since we found a dose-dependant increase of their density, but the same affinity. The highest dose of fentanyl (12.8 µg.kg-1) caused a statistically significant increase of the density of 5-HT1A receptors that was positively associated with its analgesic effect on the terminal cortical external (+64%), internal (+69%) and piriform (+113%) frontoparietal areas, the CA1 (+111%) and DGm (+60%) regions of the hippocampus, the amygdalin nuclei PMCo (+101%) and AHiAL (+91%) and the hypo-thalamus (+127%). The analgesic effect of acute fentanyl would be explained, at least, by two mechanisms: its stimulation of opiate neurotransmission acting directly on opiate µ receptors and the increase of 5-HT levels at the central nervous system and the up-regulation of 5-HT1A receptors at the terminal brain areas, hence facilitating the stimulation of such receptors. Since the post-synaptic 5-HT1A receptors act as heteroreceptors that inhibit non-serotoninergic neurons and cause neuronal hyperpolarization, fentanyl would facilitate the stimulation of these receptors and would inhibit the neuronal activity in all such terminal areas, preventing the transmission of the nociceptive stimulus. This would explain the reduced analgesic effect of opiate µ agonists that is observed with selective 5-HT1A antagonists and the increased analgesic effect observed when µ agonists and other drugs capable of increasing 5-HT levels, such as ISRS, are co-administered. Further studies are required in order to accurately determine the mechanism through which the stimulation of µ receptors cause up-regulation of post-synaptic 5-HT1A receptors and the role of each brain area in the perception of the nociceptive stimulus. © 2004 Sociedad Española del Dolor. Published by Arán Ediciones, S.L. Key words: µ receptor. 5-HT1A receptor. Synergies. Fentanyl. Analgesia. | RESUMEN Los agonistas 5-HT1A presentan efecto analgésico. El efecto analgésico de los agonistas µ puede ser bloqueado por antagonistas selectivos 5-HT1A. Para determinar el mecanismo de producción del sinergismo observado entre los receptores µ y serotoninérgico 5-HT1A en relación con su efecto antinociceptivo, determinamos el efecto analgésico de fentanilo tras estímulo nociceptivo de tipo térmico y mecánico en la rata relacionándolo con la afinidad y la densidad máxima de los receptores 5-HT1A de trece áreas cerebrales mediante técnicas de autorradiografía. Fentanilo presentó un efecto analgésico dosis y tiempo dependiente ante los dos estímulos nociceptivos. Paralelamente a la aparición del efecto analgésico, fentanilo originó una regulación a la alta de los receptores 5-HT1A al incrementar de forma dosis-dependiente su densidad sin modificar su afinidad. La dosis mayor de fentanilo (12,8 µg.kg-1) originó un incremento de la densidad de los receptores 5-HT1A estadísticamente significativo y que se correlacionó de forma positiva con su efecto analgésico en las áreas terminales corticales fronto-parietal externa (+64%), interna (+69%) y piriforme (+113%), las regiones del hipocampo CA1 (+111%) y DGm (+60%), los núcleos amigdalinos PMCo (+101%) y AHiAL (+91%) y el hipotálamo (+127%). El efecto analgésico de fentanilo en tratamiento agudo se explicaría, al menos, por dos mecanismos. Su capacidad de estimular la neurotransmisión opiácea actuando directamente sobre los receptores opiáceos µ. Y porque, al incrementar los niveles de 5-HT a nivel central y al regular a la alta los receptores 5-HT1A de zonas cerebrales terminales, se facilitaría la estimulación de estos receptores. Dado que los receptores 5-HT1A postsinápticos actúan como heteroreceptores de efecto inhibidor sobre neuronas no serotoninérgicas originando una hiperpolarización neuronal, fentanilo, al facilitar el estímulo de estos receptores originaría una inhibición de la actividad neuronal en todas estas áreas terminales impidiendo la transmisión del estímulo nociceptivo. Esto explicaría la disminución del efecto analgésico de los agonistas opiáceos µ que originan los antagonistas selectivos 5-HT1A y el mayor efecto analgésico observado al coadministrar agonistas m y fármacos capaces de incrementar los niveles de 5-HT como los ISRS. Se necesitan estudios posteriores que determinen con exactitud el mecanismo por el que el estímulo de los receptores µ origina la regulación a la alta de los receptores 5-HT1A postsinápticos y el papel de cada una de las áreas cerebrales en la percepción del estímulo nociceptivo. © 2004 Sociedad Española del Dolor. Publicado por Arán Ediciones, S.L. Palabras clave: Receptor µ. Receptor 5-HT1A. Sinergismo. Fentanilo. Analgesia.
|
1 Departamento de Farmacología y Terapeútica Clínica. Faculta de Medicina
2 Servicio de Anestesia, Reanimación y Tratamiento del Dolor. Hospital Carlos Haya
3 Servicio de Anestesia, Reanimación y Tratamiento del Dolor. Hospital Clínico Universitario. Málaga
Recibido: 23-10-03.
Aceptado: 30-04-04.
INTRODUCCIÓN
Los sistemas de neurotransmisión opiáceo y serotoninérgico a través de los receptores opiáceos µ y serotoninérgico 5-HT1A aparecen interrelacionados en diversas publicaciones. Estas interrelaciones se han descrito tanto a nivel central como periférico, afectando a la percepción del dolor, y a la presentación del efecto analgésico o de reacciones adversas de las asociaciones de diversos tipos de fármacos.
En este sentido, se ha descrito que la analgesia originada por la inyección intratecal de 5-HT aparece mediada por receptores µ (1). Los antidepresivos amitriptilina y desimipramina inducen su efecto analgésico porque incrementan los niveles de serotonina (5-HT) y porque, además pueden realizar un efecto de estimulación de los receptores medulares serotoninérgicos 5-HT1A y opiáceos µ (2-6). La co-administración de agonistas opiáceos m con inhibidores de la moanoamino oxidasa (IMAOs) o con inhibidores selectivos de la receptación de serotonina (ISRSs) puede inducir la aparición de un síndrome serotoninérgico (7).
En un trabajo previo observamos que fentanilo originaba una regulación a la alta dosis dependiente de los receptores 5-HT1A a nivel de hipocampo y de la amígdala de cerebro de rata (8). Como se ha comprobado que los agonistas 5-HT1A presentan efecto analgésico per se (9), y como la analgesia originada por los análogos del fentanilo, agonistas selectivos totales µ , puede ser bloqueada por antagonistas selectivos de los 5-HT1A carentes de efecto sobre los receptores µ(10), nos planteamos este trabajo con el objeto de determinar la posible existencia de un sinergismo entre los receptores opiáceo µ y serotoninérgico 5-HT1A en relación con su efecto analgésico.
Para ello, inicialmente realizamos curvas dosis-efecto y tiempo-efecto de fentanilo en relación con su efecto analgésico ante estímulo nociceptivo de tipo mecánico y térmico para determinar la dosis óptima a utilizar, el tiempo que debía transcurrir tras la inyección de fentanilo y el tipo de test a utilizar para poder cuantificar el efecto anti-nociceptivo del fármaco de forma objetiva y fácilmente cuantificable en la rata. Posteriormente cuantificamos la afinidad y la densidad máxima de los receptores 5-HT1A de trece áreas cerebrales mediante técnicas de autorradiografía tras el tratamiento con la dosis seleccionada de fentanilo y determinamos sus relaciones con el efecto analgésico del fármaco en el test de Randall Selitto.
MATERIAL Y MÉTODOS
El estudio se realizó en ratas de la cepa Wistar, machos (n=24, 6 meses, 335±5,4 g) (IFFA-CREDO-Charles River, Francia) estabuladas en grupos de 3 por caja en condiciones estándares con temperatura de 21±2 °C, ciclos de luz:oscuridad de 12:12 h, inicio del ciclo luminoso a las 8:00 h, comida y agua “ad libitum” y tiempo de acondicionamiento al estabulario 3 semanas. Los animales fueron inyectados intraperitonealmente (i.p.) con una dosis única de fentanilo de 3,2 µg.kg-1, 6,4 µg.kg-1 o 12,8 µg.kg-1, o con solución salina, respectivamente. La estabulación y técnicas de manejo de los animales se realizaron de acuerdo con la guía de Principios de Cuidados y Utilización de Animales de la Asociación Americana de Fisiología, con la Directiva de la Unión Europea 86/609 sobre utilización de animales con fines científicos y tras la revisión y aprobación del protocolo experimental por parte del Comité Local de Investigación Animal.
El efecto analgésico de las diferentes dosis de fentanilo se cuantificó transcurridos 1, 15 y 30 minutos de la inyección. Se determinó el efecto analgésico del fármaco ante dos tipos de estímulo nociceptivo la presión mecánica y la temperatura.
La analgesia ante nocicepción por presión mecánica de determinó mediante el test de Randall Selitto (11,12) utilizando el analgesímetro de presión en la pata posterior del animal de Ugo Basile. En este test, una pieza cónica acabada en punta roma ejerce una presión que se va incrementando de forma lineal sobre la región central de la pata posterior derecha del animal. El incremento de la presión se produce al incrementar el desplazamiento de un peso a través de una barra escalada en centímetros sobre la que realiza un efecto de palanca que se transmite a la extremidad del animal. El animal es sujetado durante todo el test de forma que puede mover libremente la pata. Cuando el animal experimenta dolor, como consecuencia del incremento de la presión, desarrolla una respuesta de retirada de la pata. Se cuantificó la presión máxima soportada por el animal antes de retirar la pata, expresada en cm (1 cm = 30 g de fuerza).
La analgesia ante nocicepción por estímulo térmico se determinó introduciendo la cola del animal en agua a temperatura de 60 ºC (13,14). El animal es sujetado en este test de forma que puede mover libremente la cola. Cuando el animal experimenta dolor, como consecuencia de la elevada temperatura del agua, desarrolla una respuesta de retirada de la cola. Se cuantificó el tiempo se reacción o tiempo que el animal tardó en retirar la cola, expresado en centésimas de segundo.
Dado que las respuestas de los animales ante los dos estímulos nociceptivos suponían un comportamiento motor y dado que los opiáceos originan un estado de sedación, se realizó el test de Open field o de campo abierto con objeto de cuantificar la función motora-exploratoria de los animales y descartar que la falta de respuesta ante el estímulo nociceptivo tras el tratamiento con fentanilo se debiera a un efecto sedante capaz de disminuir la función motora de los animales y no a un efecto analgésico. En este test el animal fue introducido en el centro una la superficie dodecaédrica de 85 cm de diámetro, de 42 cm de alto, dividida en 6 áreas de igual superficie, donde se le dejó deambular libremente durante 10 minutos (15,16). Este test se realizó en completa oscuridad de la habitación con la excepción de una única fuente lumínica de 100 watios, sujeta iluminando el área central de la superficie del campo abierto. Se cuantificaron el número de cambios de área, el número de estereotipias y el número de bolos fecales emitidos por el animal. Como en las pruebas previas no habíamos observado afectación de la función motora del animal de forma inmediata a la administración de fentanilo, este test se realizó sólo en una ocasión, antes de la tercera determinación del efecto analgésico.
Los receptores 5-HT1A fueron caracterizados mediante estudios de saturación con 3H-8OH-DPAT, agonista selectivo de los receptores 5-HT1A, utilizando técnicas de autorradiografía. Para ello, una vez cuantificado el efecto analgésico, los animales fueron inmediatamente sacrificados por decapitación y sus cerebros diseccionados rápidamente y congelados mediante inmersión en isopentano enfriado a -40 ºC en nieve carbónica. Posteriormente fueron conservados a -70 ºC hasta su utilización. Se realizaron posteriormente secciones coronales de 14 µm a partir de -3,6 mm desde Bregma que se fijaron en portas gelatinizados. Se realizó la identificación neuroanatómica de las secciones mediante comprobación histológica tras tinción con violeta de cresil utilizando el atlas de Paxinos y Watson. El radioligando 3H-8OH-DPAT (actividad específica 137 Ci.mmol-1) se utilizó en rango de concentraciones de 0,005-10 nM. Las secciones se incubaron en tampón Tris-ClH 50 mM, Cl2Ca 4 mM, ácido ascórbico 0,1% y pargilina 10 µM (pH 7,6) durante 60 minutos a temperatura ambiente. La unión no específica se determinó en presencia de 5-HT 10 mM. Tras la incubación, las secciones fueron lavadas dos veces durante 5 minutos en tampón Tris-ClH 50 mM, inmersas dos veces en agua bi-destilada bi-desionizada a 4 ºC y secadas en corriente de aire frío. Posteriormente las secciones fueron expuestas a un film sensible al tritio (3H) (3H-hyper-film) durante 6 semanas a 4 ºC (17). La unión de 3H-8OH-DPAT en corteza fronto-parietal, áreas límbicas, amígdala e hipotálamo fue cuantificada de los autorradiogramas mediante un analizador de imágenes computarizado acoplado a una vídeo-cámara con macro-objetivo. Se determinó la densidad de receptores en trece áreas. A nivel cortical en córtex fronto-parietal externo (Cort-1), córtex fronto-parietal interno (Cort-2) y córtex piriforme (C Pir). A nivel límbico en las regiones del hipocampo CA1 (CA1), CA2 (CA2), CA3 (CA3), giro dentado medial (DGm) y giro dentado lateral (DGl), en la amígdala considerada en su totalidad (AMI) y en los núcleos amigdalinos posteromedial (PMCo) y anterolateral (AHiAL), en el hipotálamo considerado en su totalidad (Hipotal) y en el núcleo talámico medio dorsal (DM). La conversión de los valores de densidad óptica de los autorradiogramas en fmol.mg-1 de proteínas se realizó a partir de los mostrados por micro-escalas poliméricas marcadas con 3H prefabricadas para autorradiografía (3H-micro-scales). El análisis de las curvas de saturación se realizó utilizando el software para análisis de regresión no lineal iterativo del programa Graph-Pad PRISMA. Se determinaron la afinidad (KD) y la densidad máxima de receptores (Bmax). 3H-8OH-DPAT, 3H-hyper-film y 3H-micro-scales fueron adquiridos de Amersham (Amersham, Buckinghamshire, U.K.) y el resto de los reactivos fueron adquiridos de Sigma (Sigma Chemical Co, St Louis, MO, EE.UU.).
Los datos aparecen expresados como el valor medio ± s.e.m. de n valores. El análisis estadístico de los datos se realizó mediante ANOVA de una y de dos vías y aplicación posterior del test de Bonferroni para determinar el grado de significación estadística entre grupos, utilizando el software del programa SPSS 11.0.
RESULTADOS
Como cabía esperar de la experiencia clínica, fentanilo presentó un efecto analgésico dosis y tiempo dependiente tanto en el test de Randall Selitto como tras el estímulo nociceptivo de tipo térmico (Figs. 1 y 2). El test de Randall Selitto, menos agresivo para el animal, fue el que permitió observar el efecto analgésico con más claridad. El efecto analgésico de fentanilo en este test fue muy rápido (Fig. 1) mostrando efecto manifiesto y estadísticamente significativo a partir de los 15 minutos de la inyección de las dos dosis mayores utilizadas en el estudio que se incrementó cuando el test de realizó a los 30 minutos a las tres dosis estudiadas. Un patrón de comportamiento similar se observa tras la estimulación nociceptiva de tipo térmico. En este caso, la dosis mayor fue la que alcanzó el efecto analgésico estadísticamente significativo a partir de los 15 minutos (Fig. 2). La bibliografía aporta numerosas citas en relación con el hecho de que el efecto analgésico originado por un fármaco depende del tipo de test de analgesia utilizado para cuantificarlo. Pero con independencia del menor efecto analgésico detectado frente al estímulo nociceptivo de tipo térmico en relación con el estímulo nociceptivo de tipo mecánico, es de destacar que observamos, como cabía esperar, la existencia de una correlación positiva entre el efecto analgésico originado por las diferentes dosis de fentanilo frente al grupo control en ambos tests, en el sentido de que los animales que presentaban la mayor analgesia ante el estímulo térmico, presentaban también la mayor analgesia ante el estímulo mecánico (coeficiente de correlación de Pearson 0,585, p = 0,001). Esto supone que con independencia de la mayor o menor sensibilidad para mostrar el efecto analgésico de los fármacos en función del tipo de test, que si el efecto se puede detectar, su forma de presentación y evolución tras ambos tipos de estímulo nociceptivo es similar.


Ninguna de las dosis de fentanilo utilizadas modificó ninguna de las variables estudiadas en relación con la capacidad motora de los animales respecto al grupo control (Fig. 3). Se puede afirmar por tanto que las respuestas desarrollas por los animales en ambos tests de analgesia se deben al efecto analgésico del fármaco y no a un posible efecto sedante o capaz de disminuir la función motora.

Al analizar la unión de 3H-8OH-DPAT en las diferentes áreas se observó en todos los casos la existencia de unión saturable a una única población de sitios de unión, como mostraban los coeficientes de Hill próximos a la unidad en todas las áreas y con las tres dosis estudiadas. Paralelamente a la aparición del efecto analgésico, fentanilo originó un incremento dosis-dependiente de la Bmax de los receptores 5-HT1A (Tabla I) sin originar modificaciones en la KD de estos receptores que supongan cambios funcionalmente destacables en su afinidad (Tabla II). Parece por tanto, que fentanilo, en tratamiento agudo induce una regulación a la alta por incremento de Bmax de los receptores 5-HT1A que aparece de forma manifiesta en todas las áreas estudiadas y que alcanzó la significación estadística en relación con todas las áreas corticales (Cort-1, Cort-2, C Pir), las regiones CA1, CA3, DGm y DGl del hipocampo, la amígdala, especialmente a nivel de los núcleos PMCo y AHiAL, y el hipotálamo en general tras la administración de la dosis mayor de fentanilo de 12,8 µg.kg-1 (Tabla I). La figura 4 muestra un autorradiograma representativo del incremento de la densidad máxima de los receptores 5-HT1A tras el tratamiento con la dosis mayor de fentanilo de 12,8 µg.kg-1 a los 30 minutos de la inyección del fármaco.


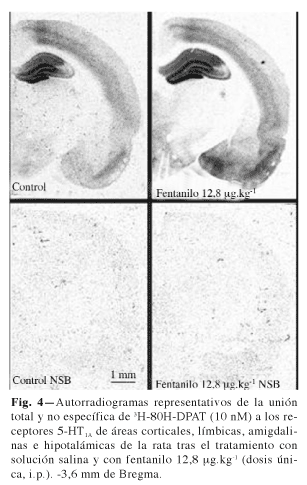
Al analizar de forma más detallada esta modificación observamos la existencia de correlaciones positivas entre el incremento del efecto analgésico de fentanilo en el test de Randall Selitto y el incremento de la Bmax de los receptores 5-HT1A detectado en varias de las áreas cerebrales estudiadas. Entre ellas destacar: CA1 (coeficiente de correlación de Pearson +0,735, p = 0,001), AHiLA (coeficiente de correlación de Pearson +0,619, p = 0,001), Cort-1 (coeficiente de correlación de Pearson +0,599, p = 0,002), Cort-2 (coeficiente de correlación de Pearson +0,563, p = 0,004), C Pir (coeficiente de correlación de Pearson +0,540, p = 0,006), DGm (coeficiente de correlación de Pearson +0,495, p = 0,014), Hipotal (coeficiente de correlación de Pearson +0,494, p = 0,014; PMCo (coeficiente de correlación de Pearson +0,457, p = 0,025), y DM (coeficiente de correlación de Pearson +0,419, p = 0,042). Aunque se observó la existencia de una tendencia a la significación, esta relación no alcanzó la significación estadística en el caso de las áreas CA2, CA3, DGl y AMI. No se detectaron tampoco correlaciones en relación con el incremento del tiempo de reacción de retirada de la cola durante la nocicepción de tipo térmico en ninguna de las áreas. Esto posiblemente se debe a la mayor dificultad que encontramos cuando realizamos este test para mostrar el efecto analgésico de fentanilo. Las modificaciones en la KD no superaron en ningún momento un factor de 2,2 y no se correlacionó en ningún caso con el efecto analgésico detectado en el test de Randall Selitto ni tras el estímulo nociceptivo térmico.
Parece, por tanto, que de alguna forma, fentanilo favorece la neurotransmisión 5-HT1A y que este efecto se relaciona, o al menos se asocia manifiestamente con su efecto analgésico en tratamiento agudo.
DISCUSIÓN
El principal resultado obtenido en este estudio es que fentanilo mostró su efecto analgésico asociado a un incremento dosis-dependiente de la densidad máxima de los receptores 5-HT1A (regulación a la alta) sin originar modificaciones en la KD de estos receptores que supongan cambios funcionalmente destacables en su afinidad (Tablas I y II y Fig. 4). Esta regulación a la alta se correlacionó positivamente con el efecto analgésico del fármaco en las tres áreas corticales estudiadas, en las áreas mediales de hipocampo (CA1 y DGm), en los principales núcleos amigdalinos (PMCo y AHiAL) y en el tálamo. El tratamiento agudo con fentanilo favorece por tanto la neurotransmisión 5-HT1A originando un posible efecto sinérgico en relación con la producción de analgesia a nivel central mediado por los receptores m y 5-HT1A. Ambos receptores junto al efecto analgésico, presentan una amplia co-localización tanto en cerebro humano como de rata, especialmente en las regiones corticales y límbicas, y comparten sistemas biológicos acoplados (7,18).
La literatura recoge que parte del efecto analgésico de los opiáceos es realizado a través de la activación de neuronas serotoninérgicas (19-22). En este sentido la administración de agonistas opiáceos µ como morfina, incrementa la síntesis, la mayor liberación y producción de metabolitos de 5-HT (23,24). Esta estimulación del turn-over de 5-HT incrementa los niveles de 5-HT en diferentes áreas del sistema nervioso central originando analgesia (18). Igualmente se ha observado que la co-administración de ISRS con agonistas opiáceos potencia el efecto analgésico de estos últimos a nivel experimental (25) y clínico (26). Este sinergismo puede originar también reacciones adversas. Este es el caso, tanto clínico como experimental, del síndrome serotoninérgico originado por la asociación de IMAOs y meperidina que aparece debido al efecto inhibidor de la recaptación de 5-HT de meperidina en presencia de un inhibidor de su metabolización. El incremento de los niveles de serotonina cerebrales originado por ambos induce la sintomatología que aparece mediada, entre otros, por receptores 5-HT1A postsinápticos (27,28).
En sentido inverso, como se mencionó anteriormente, la inyección intratecal de 5-HT origina analgesia mediada por receptores m (1), y se ha comprobado también que la depleción experimental de 5-HT con paraclorofenilalanina a nivel central disminuye el efecto analgésico de morfina (29,30).
Ni la literatura, ni trabajos previos in vitro de nuestro grupo aportan en ningún caso la existencia de capacidad de unión de fentanilo a los receptores 5-HT1A por lo que se puede descartar un efecto directo del fármaco sobre el receptor.
Para explicar este efecto sinérgico se pueden sugerir varias posibilidades considerando la posible localización de los receptores 5-HT1A a nivel pre y postsináptico dado que sus características y funciones son diferentes. A nivel presináptico, la principal localización de los receptores 5-HT1A es en los núcleos del rafe, actuando como autorreceptores somatodendríticos inhibitorios de neuronas serotoninérgicas. La estimulación de estos receptores disminuye la concentración de 5-HT en zonas terminales cerebrales de proyección (31), mientras que la administración de antagonistas selectivos como WAY 100635, facilita la función de estas células e incrementa los niveles de 5-HT en zonas terminales (28,32). Los receptores 5-HT1A se pueden también localizar a nivel presináptico en otras áreas cerebrales, entre ellas la sustancia gris periacueductal, en la que estos receptores se comportan como heterorreceptores al localizarse en interneuronas GABAérgicas. A nivel postsináptico, los receptores 5-HT1A se localizan en neuronas no serotoninérgicas en las que actúan como heterorreceptores de efecto inhibidor. Por este motivo el incremento de los niveles de 5-HT o los agonistas como el 8-OH-DPAT producen inhibición de la actividad neuronal en todas estas áreas terminales hipocampo, córtex y otras regiones cerebrales cuando se administran mediante iontoforesis in vivo, resultando en una hiperpolarización neuronal. Este efecto inhibitorio es bloqueado por la administración de antagonistas del receptor 5-HT1A, no selectivos, como el agonista parcial buspirona, y selectivos, como WAY 100635 (28). La corteza, el hipocampo, la amígdala y el hipotálamo se consideran áreas terminales o de proyección de las neuronas serotoninérgicas del rafe.
Si el sinergismo entre los receptores m y 5-HT1A ocurriera a nivel pre-sináptico se podría originar una disminución de la función interneuronas gabaérgicas localizadas en la sustancia gris periacueductal. Estas neuronas proyectan sus axones hasta neuronas espinales secretoras de sustancia P y glutamato disminuyendo la producción-liberación de ambos neuro-transmisores de efecto pro-nociceptivo (33). En condiciones basales, estas neuronas se encuentran inhibidas por el GABA por lo que la liberación de sustancia P y glutamato se produce en función de la existencia o no de estimulación nociceptiva. La estimulación de cualquiera de estos receptores 5-HT1A y/o µ que funcionan como autorreceptores presinápticos en estas interneuronas, inhibiría la liberación de GABA a nivel de las neuronas de la sustancia gris periacueductal (34,35) quedando estas neuronas des-inhibidas. Su control inhibitorio sobre las neuronas glutamérgicas y productoras de sustancia P se incrementaría entonces originando una disminución de la producción de glutamato y de sustancia P, lo que produciría analgesia (7). A nivel presináptico, ambos receptores están acoplados a canales de potasio a través de proteínas G sensibles a toxina pertusi. La estimulación de estos receptores abriría los canales originando una inhibición por hiperpolarización neuronal, en cualquier localización (36-39). Los agonistas de estos receptores disminuirían la función neuronal y su capacidad de transmitir información. Este podría ser también el punto común sobre el que se podría producir la interacción positiva. En ambos casos, si como se ha demostrado los agonistas opiáceos originan un incremento del recambio de serotonina (23,24), cabría la posibilidad de que junto al efecto estimulador opiáceo de los receptores m, la propia serotonina endógena sintetizada y liberada en exceso por efecto de los agonistas opiáceos estimulara a los receptores 5-HT1A incrementando bien la acción inhibitoria a nivel GABAérgico bien la acción hiperpolarizante. Si unimos a este efecto el incremento de los niveles de 5-HT originado por inhibidores selectivos de la recaptación de serotonina, se podría explicar el efecto sinérgico de ambos tipos de fármacos observado tanto a nivel experimental (25) como clínico (26). Otros estudios, sin embargo, muestran relaciones opuestas entre estos receptores a nivel presináptico. En este sentido se ha comprobado que la asociación de tramadol, opiáceo agonista m e inhibidor de la recaptación de serotonina y de otras monoaminas, a pindolol, antagonista adrenérgico B1 y 5-HT1A/1B, origina una potenciación del efecto analgésico de tramadol. Por el contrario, la asociación del agonista selectivo 5-HT1A 8OH-DPAT a tramadol disminuye su efecto analgésico (40). Esto nos indica que el bloqueo pre-sináptico 5-HT1A facilita la producción de analgesia, mientras que el estímulo de estos receptores disminuiría el efecto analgésico de agonistas opiáceos µ.
Considerando la localización post-sináptica de los receptores µ y 5-HT1A, la regulación a la alta de los receptores 5-HT1A en áreas cerebrales directamente relacionadas con la percepción del dolor y con la transmisión del estímulo nociceptivo, originada por el tratamiento agudo con el agonista opiáceo fentanilo, observada en este estudio, permite contemplar otro mecanismo de acción capaz de explicar la existencia de este sinergismo. La literatura recoge que la estimulación de los receptores 5-HT1A presenta efecto analgésico a nivel central (9,41). Podríamos explicar que parte del efecto analgésico de los opiáceos se realice a través del sistema serotoninérgico, primero, por el incremento de los niveles de 5-HT que origina. Y, segundo, incrementando la densidad de los receptores 5-HT1A, que al realizar un efecto heterorreceptor inhibitorio de otros sistemas neuronales, podrían inhibir el proceso de transmisión del estímulo nociceptivo originando el efecto analgésico a nivel central.
Este efecto nos permite explicar también estudios experimentales que describen como antagonistas 5-HT1A, que no muestran afinidad por los receptores opiáceos m, pueden disminuir el efecto analgésico de morfina y de otros agonistas µ (2). En este caso, el bloqueo de los receptores 5-HT1A impediría de un lado la acción de la serotonina y de otro, la realización del control inhibitorio de neuronas no serotoninérgicas.
Igualmente, este efecto de fentanilo nos permitiría explicar también la potenciación del efecto analgésico presentado por la asociación de opiáceos con los inhibidores selectivos de la recaptación de serotonina al unirse al efecto analgésico de ambos fármacos por separado, el mayor efecto de la serotonina endógena no recaptada actuando sobre una mayor cantidad de receptores 5-HT1A post-sinápticos.
Debería considerarse también una posible participación de los receptores 5-HT1A en la aparición de tolerancia y de adicción a agonistas opiáceos m. En relación con el papel de serotonina en la aparición de adicción y de tolerancia a opiáceos, se ha demostrado que niveles altos de 5-HT parecen acelerar el desarrollo de tolerancia tras tratamientos con opiáceos, mientras que los niveles reducidos tendrían un efecto contrapuesto (42).
Como anteriormente se apuntó, los agonistas opiáceos, y de ellos morfina sobre todo, incrementan los niveles de 5-HT al facilitar su turn-over (23,24). Se ha comprobado que el tratamiento agudo con morfina origina de forma dosis-dependiente un incremento de los niveles de 5-hidroxitriptófano en hipocampo (+17-25%), córtex (+24-36%), hipotálamo (+14-25%) y estriado (+22-35%), una disminución generalizada de los niveles de 5-HT en todas las áreas cerebrales y un incremento de los niveles de 5-HIAA. Estas modificaciones se podrían deber al efecto de morfina que al incrementar la liberación de 5-HT facilitaría su metabolización (43). En este mismo trabajo se recoge como la administración de 8OH-DPAT en dosis baja y capaz de actuar preferentemente a nivel de los receptores presinápticos del rafe disminuye la síntesis de hidroxitriptófano y de 5-HIAA en todas la áreas estudiadas en los animales dependientes de morfina y con síndrome de abstinencia, revirtiendo el efecto de morfina, mientras que en animales no dependientes a morfina carece de efecto (43). Se ha comprobado que los receptores 5-HT1A presinápticos de las neuronas del núcleo dorsal de rafe realizan un control inhibitorio de la actividad de la enzima triptófano hidroxilasa, responsable directo de la síntesis de 5-hidroxitriptófano, y, a través de él, del inicio de la síntesis de serotonina (44,45). Como tras el tratamiento agudo se observa un incremento de los niveles de 5-hidroxitriptófano, cabe suponer que estos receptores no están realizando su efecto inhibitorio del sistema enzimático. Dado que la administración de 8OH-DPAT revierte este efecto, es de suponer que la falta de función de estos receptores se debe a una falta de estímulo de estos receptores por serotonina debido a una disminución de los niveles del neurotransmisor a este nivel. Los receptores, ante esta falta del neurotransmisor endógeno desarrollan un estado de incremento de su sensibilidad que se demuestra por el rápido efecto de 8OH-DPAT revirtiendo los cambios originados por morfina. Pero ni este trabajo ni otras referencias de la literatura aportan datos sobre la función de los receptores del rafe.
La literatura refiere la existencia de una regulación a la baja en los receptores 5-HT1A en hipotálamo de ratas con síndrome de abstinencia por morfina que, sin embargo, no presentaban cambios en otras áreas como hipocampo, amígdala, núcleo estriado, cerebro anterior y núcleos del puente (46). Esto podría hacernos suponer que en condiciones de tolerancia-dependencia o no habría modificaciones de los receptores 5-HT1A en zonas terminales o se podría producir una regulación a la alta de estos receptores. Pero no hemos encontrado antecedentes en la literatura que corroboren, maticen o invaliden esta afirmación. Igualmente se desconoce si el desarrollo de tolerancia-adicción a fentanilo se desarrolla en estas mismas condiciones, dada su más corta vida media en relación con morfina. No hay referencias en la literatura que describan modificaciones de los receptores 5-HT1A en áreas cerebrales específicas en relación con la dependencia a opiáceos y concretamente a fentanilo. Desconocemos igualmente, los niveles de 5-hidroxitriptófano, de 5-HT y de 5-HIAA en los animales tratados de forma aguda en este trabajo. Igualmente carecemos de datos sobre las características de los receptores 5-HT1A del rafe de los animales tratados en este estudio y también desconocemos la posible evolución de los cambios receptoriales detectados en tratamientos más prolongados que queda pendiente de estudios posteriores.
Aunque resulta mucho más atractiva la relación de la modificación de los receptores 5-HT1A con el efecto analgésico de fentanilo, sobre todo tras comprobar la existencia de correlaciones positivas entre el efecto analgésico y el incremento de la densidad de los receptores 5-HT1A en varias de las áreas cerebrales estudiadas. No podemos descartar, siendo realistas, la posibilidad de que este efecto se relacione con la aparición de tolerancia y dependencia al estímulo opiáceo. En este sentido, recordar que ya ha sido descrito el autocontrol de la expresión de genes por opiáceos tras tratamiento prolongado y que ha sido relacionado con la aparición de tolerancia, dependencia y síndrome de abstinencia (47-51). En relación con fentanilo se ha observado su capacidad de originar una autorregulación a la alta en sólo 6 horas de MOR mRNA, el RNAm que codifica la síntesis del receptor opiáceo µ (52).
Dado que los receptores µ y 5-HT1A comparten sistemas de transducción como el AMPc dependiente de protein kinasa y proteínas Gi/o acopladas a varios canales iónicos, los cuales modulan también la expresión de genes, se podría aventurar que fentanilo podría de forma directa o indirecta también modificar, en este caso activándolo, la expresión de genes relacionados con la expresión de los diferentes componentes de los receptores 5-HT1A originando de esta forma la regulación a la alta observada. En contra de esta opción está el escaso tiempo transcurrido entre la administración del medicamento y el sacrificio y determinación de los receptores del animal, sólo 30 minutos, tiempo tal vez insuficiente para que se produzca la expresión genética, sea operativa y se manifieste en forma de un incremento del número de receptores.
El análisis de estos datos y de la literatura científica deja pendiente de trabajos en vías de realización y futuros, entre otros, la caracterización del efecto de fentanilo en tratamiento crónico en relación con su posible capacidad de modificar los receptores 5-HT1A centrales. Así como de relacionar más exhaustivamente estas modificaciones con la secuencia de aparición del efecto analgésico del fármaco o con el desarrollo de tolerancia y la incidencia de dependencia y de otras reacciones adversas. Todos estas cuestiones están siendo objeto de otros trabajos o lo serán en su futuro, dentro de este grupo de investigación.
| CORRESPONDENCIA: Inmaculada Bellido Estévez Departamento de Farmacología y Terapeútica Clínica Facultad de Medicina Campus de Teatinos s/n 29071 Málaga Telf.: 952 13 15 66 Fax: 952 13 15 68 e-mail:ibellido@uma.es |
BIBLIOGRAFÍA
1. Goodchild CS, Guo Z, Freeman J, Gent JP. 5-HT spinal antinociception involves mu opioid receptors: cross tolerance and antagonist studies. Br J Anaesth 1997; 78: 563-9. [ Links ]
2. Xu W, Cui X, Han JS. Spinal serotonin IA and IC/2 receptors mediate supraspinal mu opioid induced analgesia. Neuroreport 1994; 5: 2665-8. [ Links ]
3. Valverde O, Mico JA, Maldonado R, Mellado M, et al. Participation of opioid and monoaminergic mechanisms on the antinociceptive effect induced by tricyclic antidepressants in two behavioural pain test in mice. Prog Neuropsychopharmacol Biol Psychiatry 1994; 18: 1073-92. [ Links ]
4. Sierralta F, Pinardi G, Miranda HF. Effect of p-chlorophenylalanine and alpha-methyltyrosine on the antinociceptive effect of antidepressant drugs. Pharmacol Toxicol 1995; 77: 276-80. [ Links ]
5. Codd EE, Shank RP, Schupsky JJ, Raffa RB. Serotonin and norepinephrine uptake inhibiting activity of centrally acting analgesics: structural determinants and role in antinociception. J Pharmacol Exp Ther 1995; 274: 1263-9. [ Links ]
6. Levine JD, Gordon NC, Smith R, McBryde R. Desipramine enhances opiate postoperative analgesia. Pain 1986; 27: 45-9. [ Links ]
7. Kishimoto K, Koyama S, Akaike N. Synergistic mu-opioid and 5-HT1A presynaptic inhibition of GABA release in rat periacueductal gray neurons. Neuropharmacology 2001; 41: 529-38. [ Links ]
8. Bellido I, Gómez-Luque A, Castro MJ, Márquez E, et al. Acute fentanyl up-regulates the 5-HT1A receptors in the rat brain. Pharmacologist 2002; 44 (Supl. 1): 50. [ Links ]
9. Rodgers RJ, Shepherd JK. Prevention of the analgesic consequences of social defeat in male mice by 5-HT1A anxiolitics, buspirone, gepirone and ipsapirone. Psychopharmacology (Berl) 1989; 99: 374-80. [ Links ]
10. Millan MJ, Colpaert FC. 5-hydroxytryptamine (HT)1A receptors and the tail-flick response. III Structurally diverse 5-HT1A partial agonists attenuate mu- but not kappa-opioid antinociception in mice and rats. J Pharmacol Exp Ther 1991; 256: 993-1001. [ Links ]
11. Randall LO, Selitto JJ. A method for measurement of analgesic activity on inflamed tissue. Arch Int Pharmacodyn Ther 1957; 111: 409-19. [ Links ]
12. Khasar SG, Gear RW, Levine JD. Absence of nalbuphine anti-analgesia in the rat. Neuroscience Letters 2003; 345: 165-8. [ Links ]
13. Gonzáles-Ríos F, Vlaiculescu A, Ben Natan L, Protais P, Costentin J. Dissociated effects of apomorphine on various nociceptive responses in mice. J Neural Transm 1986; 67: 87-103. [ Links ]
14. Olbrich VH, Mosing M. A comparison of the analgesic effects of caudal epidural methadone and lidocaine in the horse. Vet Anaesth Analg 2003; 30: 156-64. [ Links ]
15. Silva MT, Calil HM. Screening hallucinogenic drugs: systematic study of three behavioral tests. Psychopharmacologia 1975; 42: 163-71. [ Links ]
16. Bellido I, Gómez-Luque A, García-Carrera P, Ríus F, Sánchez de la Cuesta F. Female rats show an increased sensibility to the forced swim test depressive-like stimulus in the hippocampus and frontal cortex 5-HT1A receptors. Neurosci Lett; 350: 145-8. [ Links ]
17. Blatt GJ, Fitzgerald CM, Guptill JT, Booker AB, Kemper TL, Bauman ML. Density and distribution of hippocampal neurotransmitter receptors in autism: an autoradiographic study. J Autism Dev Disord 2001; 31: 573-43. [ Links ]
18. Sandrini M, Vitale G, Otan A, Pini L-A. The potentiation of analgesic activity of paracetamol plus morphine involves the serotonergic system in rat brain. Inflamm Res 1999; 48: 120-7. [ Links ]
19. Bineau-Thurottes M, Gedefroy F, Weil-Fugaza J, Besson JM. The effects of morphine on the potassium evoked release of tritiated 5-HT from spinal cord slices in the rat. Brain Res 1984; 291: 293-9. [ Links ]
20. Drissen B, Reiman W. Interaction of the central analgesic tramadol with the uptake and release of 5-hydroxytryptamine in vitro. Br J Pharmacol 1992; 105: 147-51. [ Links ]
21. Yang SW, Zhang ZH, Wang R, Xie Y-F, et al. Norepine-phrine and serotonin induced antinociception are blocked by naloxone with different dosages. Brain Res Bull 1994; 35: 113-7. [ Links ]
22. Roberts C, Price GW, Gaster L, Jones BJ, et al. Importance of h5-HT1B receptor selectivity for 5-HT terminal autorreceptor activity: an in vivo microdialysis study in the freely-moving guinea-pig. Neuropharmacology 1997; 36: 549-57. [ Links ]
23. García-Sevilla JA, Ahtee L, Magnusson T, Carlsson A. Opiate-receptor mediated changes in monoamine síntesis in rat brain. J Pharm Pharmacol 1978; 30: 613-21. [ Links ]
24. Tao R, Auerbach SB. Increased extracellular serotonin in rat brain after systemic or intraraphe administration of morphine. J Neurochem 1994; 63: 517-24. [ Links ]
25. Nayebi AR, Hassanpour M, Rezazadeh H. Effect of chronic and acute administration of flujoxetine and its additive effect with morphine on the behavioural response in the formalin test in rats. J Pharm Pharmacol 2001; 53: 219-25. [ Links ]
26. Erjave MK, Coda BA, Nguyen Q, Donaldson G, et al. Morphine-fluoxetine interactions in healty volunteers: analgesia and side effects. J Clin Pharmacol 2000; 40: 1286-95. [ Links ]
27. Sternbach H. The serotonin syndrome. American journal of psichiatry 1991; 148: 705-13. [ Links ]
28. Barnes NM, Sharp T. A review of central 5-HT receptors and their function. Neuropharmacology 1999; 38: 1083-152. [ Links ]
29. Bodnar RJ, Kordower JH, Wallage MM, Tamir H. Stress and morphine analgesia: alterations following p-chloro-phenylalanine. Pharmacol Biochem Behav 1981; 14: 645-51. [ Links ]
30. Taber RI, Lantranyi MB. Antagonism of the analgesic effect of opioid and non-opioid agents by p-chlorophenylalanine (PCPA). Eur J Pharmacol 1981; 75: 215-22. [ Links ]
31. Morgan D, Icker MJ. Discriminative stimulus effects of the 5HT1A agonist 8-OH-DPAT: attenuation by mu but not by kappa opioids. Psychopharmacology (Berl) 1995; 122: 336-45. [ Links ]
32. Hoyer D, Clarke DE, Fozard JR, Hartig PR, et al. International Union of pharmacological classification of receptors for 5-hydroxytryptamine (serotonin). Pharmacological Reviews 1994; 46: 157-93. [ Links ]
33. Besson JM. The neurobiology of pain. Lancet 1999; 353: 1610-5. [ Links ]
34. Vaugham CW, Ingram SL, Connor MA, Christie ML. How opioids inhibit GABA-mediated neurotransmission. Nature 1997; 390: 611-4. [ Links ]
35. Vaugham CW, Christie MJ. Presynaptic inhibitory actions of opioids on synaptic transmission in the rat periaqueductal grey in vitro. The Journal of Physiology 1997; 498: 463-72. [ Links ]
36. Schmitz D, Empson RM, Heinemann U. Serotonin reduces inhibition via 5-HT1A receptors in area CA1 of rat hippocampal slices in vitro. J Neurosci 1995; 15: 7217-25. [ Links ]
37. Cheng LL, Wang SL, Gean PW. Serotonin depresses excitatory synaptic transmission and depolarization-evoked Ca2+ influx in rat basolateral amygdala via 5-HT1A receptors. Eur J Neuroscience 1998; 10: 2163-72. [ Links ]
38. Koyama S, Kubo C, Rhee JS, Akaike N. Presynaptic serotonergic inhibition of GABAergic synaptic transmission in mechanically dissociated rat basolateral amygdala neurons. The Journal of Physiology 1999; 518: 525-38. [ Links ]
39. Jeong HJ, Han SH, Min BI, Cho YW. 5-HT1A receptor-mediated activation of G-protein-gated inwardly rectifying K+ current in rat periaqueductal gray neurons. Neuropharmacology 2001; 41: 175-85. [ Links ]
40. Rojas-Corrales MO, Ortega-Alvaro A, Gibert-Rahola J, Roca-Vinardell A, Mico JA. Pindolol, a beta-adrenoceptor blocker/5-hydroxytryptamine1A/1B antagonist, enhances the analgesic effects of tramadol. Pain 2000; 88: 119-24. [ Links ]
41. Galeotti N, Ghelardini C, Bartolini A. 5-HT1A agonist induce central cholinergic antinociception. Pharmacol Biochem Behav 1997; 57: 835-41. [ Links ]
42. Li JY, Wong CH, Huang EY, Ling YC, et al. Modulations of spinal serotonin activity affect the development of morphine tolerance. Anesthesia Analgesia 2001; 92: 1563-8. [ Links ]
43. Satre-Coll A, Esteban S, García-Sevilla JA. Supersensitivity of 5-HT1A-autoreceptors and alfa2-adrenoceptors regulating monoamine synthesis in the brain of morphine-dependent rats. Naunyn-Schmiedeberg`s Arch Pharmacol 2002; 365: 210-9. [ Links ]
44. Esteban S, Lladó J, Sastre-Coll A, García-Sevilla JA. Activation and desensitization by cyclic antidepressant drugs of alfa2-autoreceptors, alfa2-heteroreceptors and 5-HT1A autoreceptors regulating monoamine synthesis in the rat brain in vivo. Naunyn-Schmiedeberg's Arch Pharmacol 1999; 360: 135-43. [ Links ]
45. Satre-Coll A, Esteban S, García-Sevilla JA. Effects of imidazoline receptor ligands on monoamine synthesis in the rat brain in vivo. Naunyn-Schmiedeberg's Arch Pharmacol 1999; 360: 50-62. [ Links ]
46. Gulati A, Bhargava HN. Down-regulation of hypothalamic 5-HT1A receptors in morphine-abstinent rats. Eur J Pharmacol 1990; 182: 253-9. [ Links ]
47. Childers SR. Opioids receptor-coupled second messenger systems. Life Sci 1991; 48: 1991-2003. [ Links ]
48. Nestler EJ. Molecular mechanisms of drugs addiction. J Neurosci 1992; 12: 2439-50. [ Links ]
49. Avidor-Reiss T, Nevo I, Levy R, Pfeuffer T, et al. Chronic opioids treatment induces adenylyl cyclase V superactivation. J Biol Chem 1996; 271: 21309-15. [ Links ]
50. Jordan B, Devi LA. Molecular mechanism of opioids receptor signal transduction. Br J Anaesth 1998; 81: 12-9. [ Links ]
51. Harrison C, Smart D, Lambert DG. Stimulatoty effects of opioids. Br J Anaesth 1998; 81: 20-8. [ Links ]
52. Yoshikawa M, Nakayama H, Ueno S, Hirano M, et al. Chronic fentanyl treatments induce the up-regulation of mu opioid receptor mRNA in rat pheochromocytoma cells. Brain Res 2000; 859: 217-23. [ Links ]

