










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista de la Sociedad Española del Dolor
versión impresa ISSN 1134-8046
Rev. Soc. Esp. Dolor vol.19 no.6 Madrid nov./dic. 2012
Buprenorfina transdérmica (Feliben®). Nueva opción terapéutica para pacientes con dolor moderado y severo
Transdermal Buprenorphine (Feliben®): new therapeutic option for patients with moderate to severe pain
C. Tornero1, J. Herrera2, O. Molà3 y J. Galván3
1Unidad del Dolor. Servicio de Anestesiología. Hospital Clínico Universitario de Valencia. Departamento de Salud Valencia Clínico-Malvarrosa. Valencia.
2Unidad del Dolor. Servicio de Anestesiología. Hospital Universitario de Valme. Sevilla.
3Departamento Médico. Laboratorios Gebro Pharma S.A. Barcelona
Dirección para correspondencia
RESUMEN
Este artículo revisa las características generales del sistema opioide y los mecanismos de acción analgésica, que han permitido desarrollar nuevas opciones terapéuticas en el tratamiento del dolor moderado a severo, tanto de origen oncológico como no oncológico. También resume los estudios que han constituido el programa de investigación y desarrollo clínico en fases I y III de una nueva formulación galénica en parches de buprenorfina transdérmica, Feliben®, en los que se compara con otras formulaciones de referencia en analgesia. Finalmente, se revisa el papel actual que tienen estas nuevas formulaciones en el manejo del dolor moderado y severo.
Palabras clave: Opioides. Buprenorfina transdérmica. Feliben®.
ABSTRACT
This paper summarizes the general characteristics of the opioid system and analgesic mechanisms that have allowed the development of new therapeutic options in the therapy areas of moderate to severe pain of both cancer and noncancer origin. It also summarizes the studies forming together the research program and clinical development (phases I and III) of a new pharmaceutical formulation of buprenorphine transdermal patches, Feliben®, which is compared with other formulations of reference in analgesia. Finally, the current opportunity context that these new formulations have in the management of moderate to severe pain is reviewed.
Key words: Opioids. Buprenorphine transdermal. Feliben®.
Introducción
Es bien conocido el impacto que el dolor produce sobre la calidad de la vida emocional, social y laboral de las personas. En 1986 se publicó la escalera analgésica de la OMS como patrón de referencia en el manejo del dolor; lo que, sin duda, ha contribuido a homogeneizar y mejorar las decisiones clínicas frente al dolor de un paciente a partir de su intensidad y persistencia. Sin embargo, desde entonces, el arsenal terapéutico se ha ampliado notablemente con nuevas opciones de tratamiento que, de un modo u otro, ponen en cuestión la escalera de referencia. De hecho, los nuevos opioides poseen cualidades analgésicas y perfiles de seguridad tales que, además de producir menos efectos adversos, proporcionan una mayor satisfacción a los pacientes (1), cuestionando los criterios de elección de fármacos del 3er escalón respecto a cuándo deben empezar a utilizarse los analgésicos opioides más potentes. La buprenorfina es un buen ejemplo de ello. En efecto, el desarrollo de formulaciones galénicas innovadoras ha hecho posible la administración de este fármaco a dosis adecuadas, equianalgésicas, de forma graduada y exponencial; lo que, a dosis reducidas, lo convierte también en un candidato adecuado para el tratamiento de dolores moderados (2-4).
La buprenorfina, principio activo del Feliben®, es un derivado semisintético del alcaloide opiáceo tebaína, presente en el opio o jugo de la Adormidera (Papaver somniferum). Es un agonista parcial de los receptores μ de bajo peso molecular y elevada liposolubilidad (5); lo que, además de conferirle una elevada potencia analgésica, le convierte en una molécula atractiva para la aplicación transdérmica. Se comercializa también en España en comprimidos sublinguales y soluciones inyectables, con indicaciones como analgésico y para manejar situaciones de dependencia a opiáceos.
El sistema opioide
El reconocimiento de un sistema opioide propio del ser humano fue posible a partir de la demostración de la existencia de: (a) sustancias endógenas sintetizadas por el propio organismo y capaces de desarrollar una respuesta al dolor; (b) receptores específicos para las mismas; y (c), unas vías de metabolización que se comportan como un modulador de la respuesta, derivada de la activación o inhibición del propio sistema. Así, se han identificado péptidos opioides que se agrupan en tres familias: endorfinas, encefalinas y dinorfinas. Más recientemente, se han identificado nuevos péptidos como la nocipeptina y la endomorfina. Unos y otros actúan sobre tres tipos de receptores selectivos denominados, μ (mu), δ (delta) y κ (kappa); aunque, como es sabido, la mayoría de los fármacos analgésicos eficaces en clínica se unen a los receptores μ.
Los receptores μ predominan en las áreas asociadas con la percepción del dolor, tales como el área periacueductal, tálamo medio y área gris periventricular. Están involucrados en respuestas de analgesia supraespinal, depresión respiratoria, miosis, dependencia física, y euforia. Existen dos subtipos de receptores μ con características biocinéticas diferentes: los μ1 que son de alta afinidad y producen principalmente analgesia, y los μ2 que son de baja afinidad y son los responsables de la depresión respiratoria (6-11). La unión de los péptidos opioides con su receptor provoca una hiperpolarización neuronal que, en definitiva, disminuye la cantidad de información que se transmite a los centros superiores, con lo que la sensación dolorosa se verá disminuida (12).
Analgésicos opioides
Las propiedades analgésicas de la adormidera son conocidas desde hace siglos, aunque su reconocimiento científico no llegó hasta que en 1805 Sertüner aisló la morfina y la identificó como la sustancia responsable del efecto analgésico del opio (13, 14).
Este grupo de analgésicos actúa sobre receptores opioides e incluye fármacos naturales como la morfina y la codeína, derivados semisintéticos como la buprenorfina y la oxicodona, y sustancias sintéticas como tramadol, tapentadol y fentanilo. Los analgésicos opioides producen analgesia fisiológica, pues simulan la acción de las endorfinas del cuerpo, ya que se unen a los receptores opioides localizados dentro y fuera del SNC. Su principal efecto es la disminución del componente sensorial y de la respuesta afectiva al dolor, que consiguen a través de dos mecanismos de acción: (a) el bloqueo de la liberación presináptica de neurotransmisores excitatorios como la sustancia P, (neurotransmisor participante en la percepción del dolor); y (b), la desensibilización de la membrana postsináptica a la acción de la sustancia P, debida a una hiperpolarización neuronal. Se trata pues de un efecto basado en la unión físico-química entre fármaco y receptor, actuando ambos como agonistas.
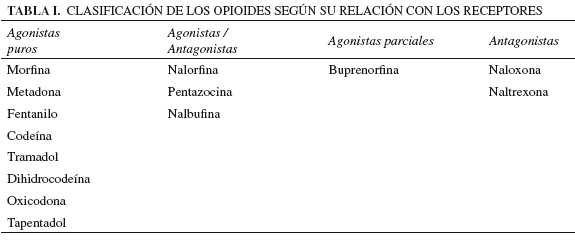
En condiciones estándar, el máximo potencial de actividad farmacológica suele corresponder a los agonistas endógenos (neurotransmisores, hormonas, etc.). No obstante, cuando se sintetizan fármacos que alcanzan una actividad biológica similar al producto endógeno, hablamos de agonistas completos o puros; y cuando no la alcanzan, de agonistas parciales. Además, en ocasiones, los fármacos pueden presentar afinidad por más de un receptor; en cuyo caso suele hablarse de fármacos duales. Cuando la afinidad por uno de los receptores genera propiedades como agonista, pero la afinidad por algún otro receptor confiere propiedades como antagonista, estamos ante fármacos mixtos denominados agonistas/antagonistas. Es el caso de algunos opioides que presentan simultáneamente afinidad por el receptor m y afinidad por otro receptor opioide, pudiéndose comportar como agonistas del primero y antagonistas del segundo (15). La tabla I clasifica a los opioides según su relación con los receptores.
Características farmacológicas de la buprenorfina
La buprenorfina es un derivado de la tebaína, lipofílico y de bajo peso molecular. Ha estado disponible como analgésico sublingual y parenteral desde 1970; y desde hace unos años se ha introducido en presentación transdérmica para el manejo del dolor crónico (16). La buprenorfina actúa como un opioide agonista parcial, con gran afinidad por el receptor μ, y como un antagonista de los receptores opioides kappa (κ). La afinidad por el receptor μ produce analgesia supraespinal, depresión respiratoria y miosis; mientras que su efecto sobre el receptor k explica cierto grado de analgesia espinal, así como sus efectos disfóricos y psicomiméticos.
Las concentraciones plasmáticas de la buprenorfina varían según la vía de administración, que asimismo determina la vida media de eliminación; de forma que en el caso de la administración endovenosa y sublingual presenta una fase rápida y otra lenta (17). La unión de la buprenorfina a las proteínas plasmáticas es muy elevada, y se metaboliza en el hígado a N-dealquilbuprenorfina (norbuprenorfina) y en metabolitos glucurónido-conjugados. Dos terceras partes del fármaco se eliminan sin metabolizar por vía biliar y las heces, pasando a la recirculación entero-hepática; en tanto que un tercio se elimina en forma no metabolizada o desalquilada por las vías urinarias; lo que clínicamente se traduce en seguridad cuando se administra a pacientes con insuficiencia renal (18).
El efecto de este fármaco es dosis o concentración dependiente hasta que se alcanza su efecto techo, a partir del cual un incremento de la dosis no conlleva incremento del efecto. Este límite mide su eficacia y es la magnitud que permite la comparación entre fármacos. En concreto, la buprenorfina presenta una analgesia similar a la morfina, produce una tasa de estreñimiento más baja (19, 20) y responde al perfil de agonista parcial. Además, la dosis a la que alcanza su efecto techo es mucho menor a la requerida por la morfina, para alcanzar el mismo nivel de efecto; es decir, la buprenorfina es un analgésico 30 veces más potente que la morfina (21). A diferencia de los agonistas m puros, como la morfina y el fentanilo, la buprenorfina carece de efecto inmunosupresor (22-24).
Nuevas formulaciones galénicas de la buprenorfina
Las formulaciones transdérmicas han supuesto un importante avance entre las opciones terapéuticas útiles para el tratamiento del dolor de intensidad moderada y severa, tanto en pacientes oncológicos como no oncológicos. La formulación transdérmica de la buprenorfina supera los problemas que plantea la farmacocinética de los opioides orales (corta duración de efecto, escasa biodisponibilidad, efectos colaterales), y parenterales (picos de concentración), favoreciendo la liberación continua a velocidad constante en la circulación sistémica, logrando una analgesia eficaz durante largos periodos, y reduciendo la frecuencia de efectos adversos.
Además, la naturaleza lipofílica de la buprenorfina y el bajo peso molecular (25) de las nuevas formulaciones permiten su administración a través de la piel sana. Con ello, se obtienen varias ventajas frente a las alternativas convencionales y se siguen las recomendaciones de la OMS para el tratamiento del dolor crónico oncológico: (a) duración de acción prolongada; (b) fluctuaciones mínimas de las concentraciones plasmáticas del analgésico, que garanticen un alivio del dolor homogéneo y a largo plazo; y (c) prevención de concentraciones plasmáticas excesivamente altas, para reducir al mínimo posible el número de efectos adversos (26-30). Asimismo, el uso de la piel como puerta de entrada en la administración sistémica del fármaco evita el metabolismo hepático de primer paso y mejora la tolerabilidad gastrointestinal. En efecto, la elevada afinidad y la baja disociación de la buprenorfina con los receptores opioides la hacen idónea para una formulación de liberación prolongada. Así, el cambio de parche se realiza cada 3 o 4 días, dependiendo del tipo utilizado. La concentración terapéutica se alcanza a las 12-24 horas y, una vez retirado el parche, la eliminación del fármaco tiene lugar aproximadamente a las 30 horas. Es por ello que en caso de sustitución del parche de buprenorfina por otro opioide se recomienda dejar un periodo de lavado de 24 horas antes de empezar a administrar el siguiente (31,32).
En resumen, el uso de la piel como puerta de entrada, en la administración sistémica del fármaco, disminuye la necesidad de administraciones repetidas de rescate (33), lo que supone una mayor comodidad para el paciente y un mejor cumplimiento de la terapia.
Por otra parte, en el ser humano la buprenorfina no muestra ningún efecto techo relevante en el intervalo posológico analgésico recomendado (25), dado que su efecto techo se expresa a dosis más altas a las empleadas para la terapia analgésica (Fig. 1). Por ello, el efecto depresor de la buprenorfina sobre el sistema respiratorio es mínimo. Además, la alta afinidad de la molécula por los receptores m le confiere características idóneas para: (a) lograr un efecto aditivo asociado a un agonista puro; y (b), ser utilizado en la rotación de opioides; puesto que provoca un menor grado de dependencia que los agonistas puros y, por tanto, un perfil de tolerabilidad más óptimo (34,35).

Las características de las nuevas formulaciones transdérmicas de buprenorfina han sido posibles gracias a las aportaciones en el diseño básico matricial de tecnología avanzada, desarrolladas para garantizar un control más seguro y preciso de la liberación del fármaco. En el mercado español hay comercializadas dos buprenorfinas transdérmicas que tienen las mismas presentaciones en parches de 35, 52,5 y 70 μg/h. La de más reciente introducción es Feliben®, que se comercializa desde enero de 2012 utilizando un sistema matricial (cuyo principio activo está incorporado directamente a una matriz de polímero), lo que permite su dosificación individual, la liberación continua del fármaco, y la eliminación del riesgo de una liberación brusca. El parche debe de ser cambiado cada 72 horas.
Desarrollo de feliben®
En el desarrollo de Feliben® se han realizado 4 ensayos clínicos de fase I, en los que han participado 64 voluntarios sanos de sexo masculino, entre 18 y 55 años, situándose la media entre los 31 y 36 años. En la tabla II se resumen por estudio los objetivos, diseño, dosificaciones y características demográficas de los pacientes. En todos los estudios se ha evaluado el perfil farmacocinético de Feliben® así como las características de seguridad y tolerabilidad. Los ensayos BUP/001/C y BUP/002/C han tenido como objetivo caracterizar el perfil farmacocinético de la nueva formulación transdérmica de parche de buprenorfina (Feliben®) respecto a la formulación de referencia Transtec® y Transtec® PRO de Grünenthal GmbH, Alemania (Transtec® PRO tiene un período de aplicación de hasta 96 horas). Además, se evaluaron y compararon las características de seguridad y tolerabilidad entre las dos formulaciones. Ambos estudios se realizaron de forma cruzada y aleatorizada, a dosis única, en régimen abierto y no controlado con placebo.

En el ensayo BUP/001/C, el periodo de tratamiento fue de 2 ciclos de 3 días, cada uno con un periodo de lavado de 6 días. Los parches presentaban un área de exposición equivalente de 25 cm2, conteniendo 20 mg de buprenorfina cada uno, estimándose la velocidad de liberación en 35 μg/h en un periodo de aplicación del parche de 72 horas, para una dosis total administrada de 2,52 mg de buprenorfina (datos de archivo).
En el ensayo BUP/002/C (36) el periodo de tratamiento, para analizar el perfil farmacocinético entre ambas formulaciones, fue de 2 ciclos de tratamiento de 7 días cada uno, con un periodo de lavado de 14 días entre cada ciclo, con parches de las mismas características descritas en el estudio anterior, para una dosis total administrada de 5,88 mg de buprenorfina.
El ensayo BUP/003/C planteó como objetivo principal la caracterización de los perfiles farmacocinéticos en voluntarios sanos de dos dosis distintas de la nueva formulación transdérmica de buprenorfina (Feliben®), investigando la proporcionalidad de ambas dosis y sus perfiles de seguridad y tolerabilidad. Las dosis estudiadas correspondían a un parche transdérmico de 25 cm2 de superficie de exposición, con 20 mg de buprenorfina y una velocidad de liberación de 35 μg/h; y a otro parche de 50 cm2, con 40 mg de buprenorfina y una velocidad de liberación estimada en 70 μg/h (datos de archivo).
Para finalizar la fase I se realizó el ensayo BUP/004/C, con el objetivo de analizar el perfil farmacocinético tras la administración de múltiples dosis de la nueva formulación de buprenorfina transdérmica (Feliben®), evaluando secundariamente su perfil de seguridad y tolerabilidad. La pauta experimental propuesta en el diseño del estudio incluía 3 ciclos consecutivos de 3 días de duración cada uno. En cada ciclo se utilizaba un parche de 25 cm2 con 20 mg de buprenorfina, con una velocidad de liberación de 35 μg/h para la administración total de 7,56 mg de buprenorfina (datos de archivo).
Los resultados del estudio BUP/001/C muestran que Feliben® alcanzó la concentración máxima en plasma antes que Transtec®. La buprenorfina se absorbió de forma más rápida con Feliben®, con una tmax inferior a Transtec® (30,3 ± 177; 16,9 h para Feliben® vs 63,6 ± 22,7 h para Transtec®), manteniéndose aproximadamente en ambas formulaciones el t1/2 (17,6 ± 177; 6,4 h en Feliben® vs 20,4 ± 20,3 h en Transtec®) (Tabla III).

Por otra parte, los resultados del estudio BUP/002/C muestran que Feliben® se libera de forma más rápida que Transtec® PRO. La concentración de Feliben® aumentó rápidamente y alcanzó su máximo valor después de 24 horas tras la aplicación del parche, disminuyendo después lentamente hasta las 168 horas, mientras que Transtec® PRO alcanzó su concentración máxima después de las 96 horas (Fig. 2), disminuyendo igualmente su concentración lentamente a partir de este momento.

En ambos estudios las dos formulaciones mostraron concentraciones máximas en plasma (Cmax) semejantes y el grado de exposición fue también similar. En cambio, tal como refleja la Fig. 2, Feliben® alcanzó la concentración máxima en un tiempo (tmax) significativamente menor que Transtec® PRO.
En el ensayo BUP/003/C los perfiles de concentración media en función del tiempo mostraron una tendencia paralela proporcional a la dosis, con concentraciones máximas a las 24 horas después de la aplicación de los parches; y a continuación, los perfiles se redujeron poco a poco hasta la retirada del parche transdérmico a las 72 horas.
El ensayo BUP/004/C mostró que al aplicar 3 parches consecutivos durante 3 días cada uno (9 días en total) sin periodo de lavado entre cada aplicación, se alcanzan los niveles máximos de buprenorfina a las 24 horas después de la aplicación de cada parche; en tanto que los niveles mínimos se alcanzan a las 72 horas después de cada aplicación y antes de la aplicación del siguiente parche. La Cmax absoluta de buprenorfina se obtuvo a las 24 horas después de la aplicación del tercer parche. Por otra parte, la eliminación en plasma después de retirar el tercer parche fue lenta, debido a la larga vida media de este fármaco (aproximadamente 30 ± 10 horas).
En los cuatro ensayos clínicos realizados, las reacciones adversas observadas con mayor frecuencia tras la administración de Feliben® fueron dolor de cabeza de intensidad leve-moderada, náuseas y vómitos de intensidad moderada; así como mareos, estreñimiento, cansancio, y vértigo rotatorio. Merece comentarse que todas las reacciones adversas se resolvieron antes de la finalización de los respectivos estudios y ninguna se consideró de intensidad severa.
Las reacciones adversas locales más frecuentes en el lugar de aplicación fueron eritema y prurito. En los estudios BUP/001/C y BUP/002/C, en los que se comparó Feliben® con Transtec® y Transtec PRO®, respectivamente, no se produjeron diferencias significativas entre los tratamientos en cuanto a reacciones adversas locales ni sistémicas.
El desarrollo de Feliben® incluyó también un ensayo clínico de Fase III (37) (BUP/005/C), que se realizó entre septiembre de 2005 y diciembre de 2006, en 26 centros de 3 países europeos, con el propósito de demostrar la no inferioridad terapéutica de Feliben® respecto a Transtec®.
De los 201 pacientes inicialmente seleccionados con dolor oncológico crónico y severo, inadecuadamente controlado con otros analgésicos, fueron aleatorizados 165 (80 pacientes al grupo de Transtec® y 85 al de Feliben®). La variable principal del estudio se evaluó mediante el cambio en la valoración del dolor, desde el momento inicial hasta el momento final, durante la fase de tratamiento. Los pacientes registraron su evaluación del dolor dos veces al día usando una escala Likert verbal de 5 puntos, realizándose la media de los dos tests. El margen de no inferioridad se estipuló en -0,25. Además, los datos obtenidos con la escala de Likert fueron confirmados con otro cuestionario, el Brief Pain Inventory (BPI), que se cumplimentó en cada visita de la fase de tratamiento activo.
En ambos grupos se redujo en más de un 30% de media la percepción del dolor, tanto en periodo diurno como nocturno, no hallándose diferencias estadísticamente significativas entre los dos grupos de tratamiento; de modo que la no inferioridad de Feliben® respecto a Transtec® quedó evidenciada. Asimismo, la cantidad de medicación de rescate necesaria durante la fase de tratamiento activo fue comparable entre los dos brazos y en ambos grupos se mejoró la duración del sueño ininterrumpido, confirmando los resultados de estudios previos con Transtec®. Además, los dos parches mostraron una buena adhesividad en el 80% o más de todos los pacientes, aunque en este aspecto Feliben® resultó ser algo mejor que Transtec®, si bien las diferencias observadas no fueron estadísticamente significativas. Asimismo, tampoco se evidenciaron diferencias significativas respecto a la frecuencia de efectos adversos entre los dos grupos de tratamiento. De hecho, se observaron 9 acontecimientos adversos graves, evaluados en todos los casos como "no relacionados con los fármacos objeto del estudio", en 9 pacientes con una distribución similar entre ambos grupos experimentales (Transtec® n = 5, Feliben® n = 4).
Ventajas de la buprenorfina transdérmica
Los beneficios del tratamiento analgésico con formulaciones opioides transdérmicas, en pacientes con dolor de origen oncológico, han sido demostrados por la reducción del consumo de comprimidos sublinguales de tramadol, de entre un 50 y un 70%, junto a una mejora de los perfiles de percepción del dolor y a un incremento de la duración del sueño no interrumpido por el dolor, además de una mejora en la calidad de vida, estado físico, bienestar subjetivo, actividad cotidiana y salud global percibida (38). Asimismo, en una serie de casos publicados se confirmó la utilidad del tratamiento con buprenorfina transdérmica en pacientes con dolor por cáncer renal y cáncer metastásico de próstata y de mama, demostrándose también la eficacia a largo plazo en el tratamiento del dolor oncológico avanzado y la posibilidad de incluir a la buprenorfina en la rotación con otros opioides (39).
Por otra parte, la buprenorfina transdérmica también ha permitido mejorar el abordaje del dolor crónico no oncológico, dado que en estos casos, con frecuencia, la intensidad del dolor y su persistencia hacen aconsejable la utilización de fármacos opioides de apoyo a la analgesia, propia del 2.o escalón de la OMS.
En cuanto al manejo de pacientes con problemas respiratorios, el tratamiento con buprenorfina ha evidenciado, en estudios comparativos con agonistas puros como el fentanilo, un riesgo significativamente menor de inducir depresión respiratoria (40).
Respecto a su administración en pacientes con insuficiencia renal conviene destacar que la buprenorfina se excreta principalmente (aproximadamente el 80-90%) por vía biliar, por lo que su excreción no se ve afectada en pacientes con deterioro de la función renal. También merece señalarse que en pacientes ancianos no se requiere un ajuste de dosis. En cambio, en pacientes con insuficiencia hepática se debe tener en cuenta que la vía metabólica principal de la buprenorfina es la glucurono-conjugación (41), por lo que la intensidad y la duración de acción de la buprenorfina transdérmica pueden verse afectadas en estos pacientes y, por ello, es necesario monitorizarlos estrechamente.
La buprenorfina transdérmica debe adaptarse a las necesidades iniciales del paciente en lo que se refiere a intensidad del dolor (utilizándose la dosis más baja posible que consiga el alivio del dolor), tal como sucede con otros opioides. No obstante, antes de elegir la concentración del parche de buprenorfina transdérmica, en pacientes tratados previamente con opioides débiles (como tramadol) o potentes (como morfina), se deben tener en cuenta el tipo, la administración y la dosis diaria media del tratamiento previo. Para cambiar a la buprenorfina desde analgésicos del 2.o escalón se aplicará la misma norma que en los pacientes que no han recibido tratamientos previamente con opioides, esto es, comenzar con la dosis más baja de buprenorfina transdérmica y ajustar progresivamente al alza, hasta lograr el efecto deseado, teniendo siempre en cuenta el posible desarrollo de tolerancia ante el opioide previo. Para ello, después de aplicar el primer parche de buprenorfina transdérmica, se recomienda mantener durante las primeras 12-24 horas el fármaco previo, esperando a que las concentraciones plasmáticas de buprenorfina alcancen su nivel analgésico. Así, la primera evaluación del efecto analgésico solo debe hacerse después de transcurridas las primeras 24 horas. En caso necesario, se aumentará la dosis aplicada cambiando el parche por el de la concentración inmediatamente superior, ajustando la dosis individualmente hasta alcanzar la eficacia analgésica.
Conclusiones
La buprenorfina está indicada en el tratamiento del dolor crónico de moderado a intenso. A diferencia de los agonistas m puros, carece de efecto inmunosupresor, deprime en menor grado el centro respiratorio, provoca un menor grado de dependencia, presenta una tasa de estreñimiento más baja (19,20), y su excreción no se ve afectada en pacientes con deterioro de la función renal. Estas evidencias convierten a la buprenorfina en una molécula clave para el tratamiento a largo plazo de pacientes con dolor moderado y severo. En efecto, aunque se clasifica como analgésico de tercer escalón en la escalera analgésica de la OMS, se ha propuesto como analgésico intermedio entre el escalón 2 y el 3, tanto para tratar el dolor oncológico como el no oncológico (42).
La nueva formulación transdérmica de Feliben® ha demostrado, en los estudios de fase I, una cinética de absorción de la buprenorfina más rápida que Transtec®, optimizando los perfiles conocidos hasta la fecha. Asimismo, el ensayo clínico en fase III ha permitido demostrar la equivalencia del perfil de eficacia y seguridad de Feliben® con el referente disponible en España (Transtec®). El parche de Feliben® libera el fármaco de forma continua a velocidad constante, por lo que se consiguen concentraciones plasmáticas eficaces durante periodos prolongados de tiempo. La concentración terapéutica se alcanza a las 12-24 horas y la eliminación del fármaco tiene lugar, aproximadamente, a las 30 horas tras la retirada del parche. De esta manera Feliben® permite una cómoda dosificación cada 72 horas, lo que puede favorecer un mejor cumplimiento terapéutico (43) que, junto a sus características de eficacia y tolerabilidad, le otorgan un perfil adecuado para el tratamiento analgésico opioide continuado, tan necesario en el paciente que sufre dolor crónico.
Bibliografía
1. Marinangeli F, Ciccozzi A, Leonardis M, et al. Use of strong opioids in advanced cancer pain: a randomized trial. J Pain Symptom Manage. 2004 May;27(5):409-16. [ Links ]
2. Romero J, Gálvez R, Ruiz S. ¿Se sostiene la Escalera Analgésica de la OMS? Rev Soc Esp Dolor. 2008;1:1-4. [ Links ]
3. Eisenberg E, Berkey CS, Carr DB, et al. Efficacy and safety of nonsteroidal antiinflammatory drugs for cancer pain: a meta-analysis. J Clin Oncol. 1994 Dec;12(12):2756-65. [ Links ]
4. McNicol E, Strassels S, Goudas L, et al. Nonsteroidal anti-inflammatory drugs, alone or combined with opioids, for cancer pain: a systematic review. J Clin Oncol. 2004 May 15;22(10):1975-92. [ Links ]
5. Sociedad Científica Española de estudios sobre el Alcohol el Alcoholismo y las otras toxicomanías. Guía para el tratamiento de la adicción a opiáceos con buprenorfina/naloxona. Guías clínicas SOCIDROGALCOHOL basadas en la evidencia científica; 2010. [ Links ]
6. Di Marzo V, Fontana A, Cadas H, et al. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994 Dec 15;372(6507):686-91. [ Links ]
7. Pertwee RG. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol Ther. 1997;74(2):129-80. [ Links ]
8. Brown AJ. Novel cannabinoid receptors. Br J Pharmacol. 2007 Nov;152(5):567-75. [ Links ]
9. Kozak KR, Prusakiewicz JJ, Marnett LJ. Oxidative metabolism of endocannabinoids by COX-2. Curr Pharm Des. 2004;10(6):659-67. [ Links ]
10. Ruzicka BB, Fox CA, Thompson RC, et al. Primary astroglial cultures derived from several rat brain regions differentially express mu, delta and kappa opioid receptor mRNA. Brain Res Mol Brain Res. 1995 Dec 28;34(2):209-20. [ Links ]
11. Massi P, Valenti M, Bolognini D, et al. Expression and function of the endocannabinoid system in glial cells. Curr Pharm Des. 2008;14(23):2289-98. [ Links ]
12. Stein C, Clark JD, Oh U, et al. Peripheral mechanisms of pain and analgesia. Brain Res Rev. 2009 Apr;60(1):90-113. [ Links ]
13. Gutstein HB, Akil H. Analgésicos opioides. En: Brunton Ll LJ, Parker KL, (editores). Las bases farmacológicas de la terapéutica. México DF: Editorial McGraw Hill; 2007. p. 547-89. [ Links ]
14. Gaoni Y, Mechoulam R. Isolation, structure, and partial synthesis of an active constituent of hashish. J Am Chem Soc. 1964;86(8):1646-7. [ Links ]
15. Grupo de trabajo de la Guía de Práctica Clínica sobre Cuidados Paliativos. Guía de Práctica Clínica sobre Cuidados Paliativos. Madrid: Plan Nacional para el SNS del MSC. Agencia de Evaluación de Tecnologías Sanitarias del País Vasco; 2008. [ Links ]
16. Evans HC, Easthope SE. Transdermal buprenorphine. Drugs. 2003;63(19):1999-2010; discussion 1-2. [ Links ]
17. Bullingham RE, McQuay HJ, Moore A, et al. Buprenorphine kinetics. Clin Pharmacol Ther. 1980 Nov;28(5):667-72. [ Links ]
18. Heel RC, Brogden RN, Speight TM, et al. Buprenorphine: a review of its pharmacological properties and therapeutic efficacy. Drugs. 1979 Feb;17(2):81-110. [ Links ]
19. Griessinger N, Sittl R, Likar R. Transdermal buprenorphine in clinical practice--a post-marketing surveillance study in 13,179 patients. Curr Med Res Opin. 2005 Aug;21(8):1147-56. [ Links ]
20. Soto Niño MC. Buprenorfina en el manejo de dolor por cáncer. Rev Colomb Cancerol. 2009;13(2):99-104. [ Links ]
21. Sigmon SC, Wong CJ, Chausmer AL, et al. Evaluation of an injection depot formulation of buprenorphine: placebo comparison. Addiction. 2004 Nov;99(11):1439-49. [ Links ]
22. Fecho K, Maslonek KA, Dykstra LA, et al. Evidence for sympathetic and adrenal involvement in the immunomodulatory effects of acute morphine treatment in rats. J Pharmacol Exp Ther. 1996 May;277(2):633-45. [ Links ]
23. Sacerdote P, Manfredi B, Mantegazza P, et al. Antinociceptive and immunosuppressive effects of opiate drugs: a structure-related activity study. Br J Pharmacol. 1997 Jun; 121(4):834-40. [ Links ]
24. Martucci C, Panerai AE, Sacerdote P. Chronic fentanyl or buprenorphine infusion in the mouse: similar analgesic profile but different effects on immune responses. Pain. 2004 Jul;110(1-2):385-92. [ Links ]
25. Lewis JW. Buprenorphine. Drug Alcohol Depend. 1985 Feb;14(3-4):363-72. [ Links ]
26. Bartleson JD. Evidence for and against the use of opioid analgesics for chronic nonmalignant low back pain: a review. Pain Med. 2002 Sep;3(3):260-71. [ Links ]
27. Kalso E. Route of opioid administration: does it make a difference. In: Kalso E, McQuay HJ, Wiesenfeld-Hallin Z (editors). Opioid sensitivity of chronic noncancer pain Progress in pain research and management. Seattle: IASP press; 1999. [ Links ]
28. Budd K. Experience with partial agonists in the treatment of cancer pain. In: Doyle D (editor). Opioid in the treatment of Cancer Pain. London: Royal Society of Medicine; 1990. p. 51-5. [ Links ]
29. Portenoy RK, Foley KM, Inturrisi CE. The nature of opioid responsiveness and its implications for neuropathic pain: new hypotheses derived from studies of opioid infusions. Pain. 1990 Dec;43(3):273-86. [ Links ]
30. Kogel B, Christoph T, Strassburger W, et al. Interaction of mu-opioid receptor agonists and antagonists with the analgesic effect of buprenorphine in mice. Eur J Pain. 2005 Oct;9(5):599-611. [ Links ]
31. Kress HG. Clinical update on the pharmacology, efficacy and safety of transdermal buprenorphine. Eur J Pain. 2009 Mar;13(3):219-30. [ Links ]
32. Rull M, Puig R. Manejo de buprenorfina transdérmica en pacientes que no han usado previamente opioides. Rev Soc Esp Dolor. 2006;13(2). [ Links ]
33. Plancarte R, Gutierrez H. Buprenorfina transdérmica en pacientes con dolor oncológico. Cancerología. 2006;1:253-71. [ Links ]
34. Staat PS, Johnson RE. New perspectives on the pharmacology of opioids and their use in chronic pain. Prog Anaesthesiol. 2002;16:235-49. [ Links ]
35. Zaki PA, Keith DE Jr., Brine GA, et al. Ligand-induced changes in surface mu-opioid receptor number: relationship to G protein activation? J Pharmacol Exp Ther. 2000 Mar;292(3):1127-34. [ Links ]
36. Molà O, et al. Estudio de fase I en voluntarios sanos para evaluar las propiedades farmacocinéticas del nuevo parche de buprenorfina transdérmica Feliben® en comparación con el fármaco de referencia Transtec® PRO. Póster presentado en el 38o Congreso Nacional de la SER, Zaragoza, 15-18 Mayo de 2012. Abstract en: Reumatología Clínica 2012;8 (Espec. Cong.):156. [ Links ]
37. Molà Villa O, et al. Estudio comparativo de la eficacia analgésica y la tolerabilidad de dos buprenorfinas transdérmicas en pacientes con dolor crónico severo por cáncer no controlados adecuadamente con otros analgésicos. Póster presentado en el 9.o Congreso de la Sociedad Española del Dolor. Barcelona, 6-9 junio 2012. Abstract en: Rev Soc Esp Dolor. 2012:19(supl II):42. [ Links ]
38. Muriel C, Grupo de Estudio de Opioides de la Sociedad Española de Dolor. Valoración del parche transdérmico de buprenorfina en pacientes con dolor oncológico. Rev Soc Esp Dolor. 2004;11(Supl):41-8. [ Links ]
39. Schriek P. Treatment of cancer-related pain with transdermal buprenorphine: a report of three cases. Support Care Cancer. 2004 Dec;12(12):882-4. [ Links ]
40. Dahan A, Yassen A, Bijl H, et al. Comparison of the respiratory effects of intravenous buprenorphine and fentanyl in humans and rats. Br J Anaesth. 2005 Jun;94(6):825-34. [ Links ]
41. Tegeder I, Lotsch J, Geisslinger G. Pharmacokinetics of opioids in liver disease. Clin Pharmacokinet. 1999 Jul;37 (1):17-40. [ Links ]
42. Radbruch L. A therapeutics masterclass. Eur J Palliat Care. 2003;10 (1 suppl):20-120. Grunenthal GmbH. Transtec® scientific monograph. Aachen: Gruenenthal GmbH. 2002. [ Links ]
43. Terlinden R, Stadler T. Buprenorphin in einem transdermalen therapeutischen system - Pharmakokinetische studie bei einfachapplikation. Deutscher Schmerzkongress; 2000; Hamburg. 2000. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Oriol Molà
Departamento Médico.
Laboratorios Gebro Pharma S.A.
Barcelona. España
E-mail: oriol.mola@gebro.es
Recibido: 05-07-12.
Aceptado: 13-08-12.

