










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versión impresa ISSN 1137-6627
Anales Sis San Navarra vol.31 supl.1 Pamplona 2008
Los trastornos del movimiento en urgencias
Movement disorders in the emergency department
M.E. Erro1, I. Gastón2, M.C. Navarro1
1. Servicio de Neurología. Hospital de Navarra. Pamplona.
2. Servicio de Neurología. Hospital Virgen del Camino. Pamplona.
Dirección para correspondencia
RESUMEN
Los pacientes que acuden a urgencias con un cuadro clínico en el que predomina un trastorno del movimiento de instauración aguda o subaguda suponen un porcentaje pequeño de las urgencias neurológicas. Sin embargo, su conocimiento es importante ya que en muchos de estos casos un error en el diagnóstico o tratamiento puede conllevar una importante morbilidad e incluso mortalidad. La forma clínica de presentación de estos trastornos es variada y en algunos predomina la acinesia o la rigidez mientras que en otros casos son los movimientos anormales en forma de discinesias o balismos lo que caracterizan al cuadro clínico. El tipo de trastorno del movimiento orienta hacia una determinada etiología. El consumo de determinados fármacos o tóxicos supone una de las principales causas de trastorno agudo del movimiento dentro de las que se incluyen el síndrome neuroléptico maligno y el síndrome serotoninérgico. En esta revisión se ha dedicado un apartado a las urgencias que plantea la enfermedad de Parkinson y que incluyen el síndrome parkinsonismo-hiperpirexia, la psicosis aguda y las urgencias de los pacientes con neuroestimuladores. Las distonías y corea-balismo agudas son también abordadas, y por último se dedica un apartado a las trastornos del movimiento como forma de presentación de un ictus.
Palabras clave. Síndrome neuroléptico maligno. Síndrome serotoninérgico. Urgencias en la enfermedad de Parkinson. Distonía secundaria a fármacos.
ABSTRACT
Acute or sub-acute movement disorders represent a small percentage of neurological emergencies but it is necessary to be aware of their existence because a failure in their diagnosis or treatment can result in significant morbidity and mortality. Clinical presentation of acute movement disorders can be diverse. In some cases acinesia or rigidity predominates, while others are characterized by dystonia, chorea o balism. The type of movement disorder suggest a specific aetiology. Drugs represent the most frequent etiologic factor and are the cause of neuroleptic malignant syndrome and serotoninergic syndrome. Emergencies secondary to Parkinsons disease are reviewed, including parkinsonism-hyperpirexia syndrome, acute psychosis and the emergencies derived from deep brain stimulators. Different aetiologies of acute dystonia and chorea are also covered and, finally, acute movement disorders due to stroke are reviewed.
Key words. Neuroleptic malignant syndrome. Serotonergic síndrome. Emergencies in Parkinsons disease. Drug induced dystonia.
Definición
Los trastornos del movimiento que requieren una atención en urgencias son aquellas enfermedades neurológicas de evolución aguda o subaguda en las que en la presentación clínica predomina el trastorno del movimiento y en las que un error en el diagnóstico o tratamiento puede conllevar importante morbilidad e incluso mortalidad1. Suponen un porcentaje pequeño dentro de las consultas neurológicas urgentes y pueden tener etiologías muy diversas (Tabla 1) dentro de las que hay que destacar aquéllos secundarios a fármacos (Tabla 2). Se pueden clasificar según la forma de presentación en los que cursan con rigidez, parkinsonismo, distonía, corea o balismo y mioclonías2.


La rigidez es la forma de presentación del síndrome neuroléptico maligno, el síndrome serotoninérgico, la hipertermia maligna, el síndrome de la persona rígida y la catatonia letal (Tabla 3). Además, pueden presentarse con rigidez enfermedades infecciosas como el tétanos o la rabia, la intoxicación por estricnina3, la hipocalcemia y enfermedades hereditarias como la hiperecplexia4.

Síndrome neuroléptico maligno
El síndrome neurológico maligno (SNM) es un proceso patológico inducido por fármacos con efecto antidopaminérgico, principalmente por los neurolépticos clásicos como el haloperidol o la clorpromacina. También pueden producir un SNM los antieméticos como la metoclopramida y antihistamínicos como la prometacina5. Se produce en el 0,2% de los casos tras consumo de antipsicóticos atípicos (olanzapina, clozapina, risperidona, quetiapina, zisapridona y aripiprazol) y la asociación de litio a cualquiera de ellos puede favorecer su aparición6. Es más frecuente en hombres jóvenes que utilizan preparados de liberación lenta y se favorece por la deshidratación. Se trata de una reacción idiosincrásica que aparece a las pocas semanas de iniciado el tratamiento o tras un aumento en la dosis y se caracteriza por rigidez muscular grave, alteración del nivel de conciencia y disautonomía con fiebre, sudoración, taquicardia, taquipnea y labilidad de la tensión arterial. En los análisis se observa una elevación de la creatin kinasa (CK) como consecuencia de la rabdomiolisis que se produce por la rigidez y puede provocar mioglobinuria con fallo renal secundario7.
En cuanto a la fisiopatología se barajan dos hipótesis. Un mecanismo central por bloqueo dopaminérgico en el estriado (rigidez) y en el hipotálamo (disautonomía) o un mecanismo periférico por alteración de la capacidad de contracción de la fibra muscular debido a una alteración de la membrana del retículo sarcoplásmico5.
El tratamiento consiste en suspender los neurolépticos (Tabla 4), medidas generales de soporte vital (hidratación y ventilación mecánica si fuese preciso), administración de fármacos de acción dopaminérgica como la bromocriptina a una dosis inicial de 2,5 mg /8h y aumentándola uno o dos días después a 5 mg/8h por vía oral (sonda nasogástrica) También pueden utilizarse la apomorfina subcutánea o los parches transdérmicos de rotigotina6. El dantrolene a dosis inicial de 25 mg/24h con aumentos posteriores de 25 mg/12h o 1-10 mg/Kg por vía intravenosa puede emplearse en cuadros graves. Se debe considerar la terapia electroconvulsiva en pacientes psicóticos5. Sin tratamiento se estima que la mortalidad asciende al 20% y las causas de muerte son el tromboembolismo pulmonar, la neumonía por aspiración y el fallo renal.

Síndrome serotoninérgico
El síndrome serotoninérgico (SS) es una reacción adversa a fármacos de acción serotoninérgica que puede ser potencialmente mortal. Tiene una incidencia de aproximadamente el 14-16% de los pacientes que toman sobredosis de inhibidores selectivos de la recaptación de la serotonina (ISRS) pero puede aparecer tras el consumo de otros fármacos con acción serotoninérgica (Tabla 5).

Se caracteriza clínicamente (Tabla 3) por alteración del estado mental (delirio, ansiedad, hipervigilancia, acatisia), hiperactividad autonómica (diarrea, sudoración, taquicardia, hipertermia, midriasis) y alteraciones neuromusculares (temblor, hiperreflexia, mioclonias, rigidez). El comienzo de los síntomas suele ser rápido, de minutos a horas después de tomar la medicación. Los hallazgos de laboratorio consisten en acidosis metabólica, rabdomiolisis, aumento de CK, fallo renal e incluso datos de coagulación intravascular diseminada8. El SS no es una reacción idiopática sino que se produce como consecuencia de un exceso de acción serotoninérgica en los receptores serotoninérgicos del sistema nervioso central y periféricos como consecuencia de interacciones farmacológicas o abuso voluntario como intento de suicidio9. En cuanto a la fisiopatología parece ser que es la activación de los receptores 5-HT2A la que contribuye fundamentalmente a las manifestaciones del SS10. El tratamiento (Tabla 4) consiste en retirar el fármaco precipitante, medidas de soporte vital, control de la agitación, de la hipertermia y de la inestabilidad autonómica. En algunos casos sólo con suspender el fármaco precipitante y con medidas generales los síntomas se resuelven en 24 horas. En otros casos, a las medidas previas sólo es necesario añadir el uso de benzodiacepinas. Las medidas de contención física deben evitarse. Hay que estar alerta porque pueden producirse empeoramientos clínicos rápidos. En casos moderadamente graves es necesario administrar fármacos de acción antagonista 5-HT2A como la ciproheptadina. a dosis de 4-8 mg oral de inicio seguido de una dosis de mantenimiento de hasta 8 mg/6 horas. Sólo puede administrarse por vía oral y por sonda nasogástrica. Cuando la hipertermia está por encima de 41º C puede ser necesaria la parálisis neuromuscular con agentes no despolarizantes y la intubación orotraqueal. Los antipiréticos no tienen papel en el manejo de la hipertermia en la que no interviene el hipotálamo sino la actividad muscular. La succinilcolina debe evitarse por el riesgo de arritmia que puede producir en asociación con la hipercalcemia que se produce con la rabdomiolisis. Fármacos como la bromocriptina, el propanolol o el dantrolene no se recomiendan10.
Hipertermia maligna
La hipertemia maligna es una grave enfermedad con susceptibilidad genética con carácter autosómico dominante y penetrancia variable debida a la mutación de un gen del receptor de la rianodina en el cromosoma 1911. Las mutaciones de este receptor van a provocar una alteración en la homeostasis del calcio con incremento del calcio muscular y contracción sostenida del músculo, lo que a su vez conlleva un estado de hipermetabolismo exagerado con acidosis láctica, hipercapnia e hipertermia. Se desencadena por agentes anestésicos halogenados inhalados y relajantes musculares despolarizantes. Las manifestaciones clínicas (Tabla 3) pueden aparecer en el quirófano o varias horas después de la anestesia. El signo más frecuente y precoz es la taquicardia, después aparece rigidez muscular en forma de espasmo de maseteros y taquipnea. Hay acidosis metabólica y respiratoria. La elevación de la temperatura es un signo tardío, así como las arritmias, la cianosis, la hipotensión, la hiperpotasemia y la rabdmiolisis12. El tratamiento (Tabla 4) consiste en hiperventilar al paciente con oxígeno al 100%, administrar dantroleno a dosis de 2,5 mg/kg en bolos, cada cinco minutos y luego en perfusión a dosis de 1-2 mg/kg/h, reducir la temperatura corporal con medidas físicas, tratar la acidosis con bicarbonato sódico, vigilar la diuresis y tratar las arritmias9.
Catatonia letal
La catatonia letal es un síndrome con distintas causas que suele aparecer en el contexto de enfermedades psiquiátricas y puede estar desencadenado por infecciones del sistema nervioso, hipertiroidismo, lupus, tóxicos, traumatismos, etc. Suele aparecer en personas jóvenes y su relación con el SNM no está clara. Se caracteriza (Tabla 3) por insomnio, anorexia, excitación motora catatónica, confusión, disautonomía (sudoración, taquicardia y variaciones de la tensión arterial), alucinaciones, ecolalia, ecopraxia, mirada fija, posturas fijas, mutismo, rigidez intensa y fiebre alta. En cuanto a la fisiopatología se especula que existe una afectación de la transmisión dopaminérgica. El tratamiento (Tabla 4) consiste en medidas de soporte vital junto con loracepam intravenoso y es de elección la terapia electroconvulsiva5.
Síndrome de la persona rígida
El síndrome de la persona rígina (SPR) es un cuadro que puede aparecer de forma aguda o subaguda y se caracteriza por la aparición de dolor y espasmos o contracturas de los músculos paraespinales lumbares, abdominales y de extremidades inferiores lo que produce una exagerada lordosis lumbar, que puede ocasionar fracturas, subluxaciones articulares o herniación abdominal. Se asocia a hiperreflexia generalizada. Las contracturas pueden aparecer de forma espontánea o tras desencadenantes auditivos o sensitivos y desaparecen durante el sueño. Los estudios neurofisiológicos sugieren que se trata de una actividad motora continua central. Se acepta que existe un SPR idiopático, una variante paraneoplásica, otros asociados a encefalomielitis y un síndrome focal o del miembro rígido. Al menos la mitad de los pacientes tienen anticuerpos anti-descarboxilasa del ácido glutámico (GAD) y también se asocia a anticuerpos antianfifisina y anti-gephyrina. En algunas formas de SPR se ha demostrado que la eficacia del tratamiento con inmunoglobulinas intravenosas13,14.
Urgencias en la enfermedad de parkinson
Síndrome de acinesia aguda-hiperpirexia
También denominado síndrome maligno o SNM-like. Aparece en pacientes con enfermedad de Parkinson (EP) de larga evolución que ingresan en un hospital por un proceso intercurrente o intervención quirúrgica y han realizado una reducción importante y brusca en la medicación antiparkinsoniana. Puede aparecer también en otros síndromes rígido-acinéticos diferentes a la EP. Se caracteriza (Tabla 3) por la instauración brusca de una acinesia extrema junto con aumento de temperatura corporal. Otras manifestaciones clínicas que pueden aparecer son rigidez espontánea, disminución del nivel de conciencia (desde somnolencia leve al coma), alteración autonómica con taquicardia, fluctuaciones de la tensión arterial, falta de sudoración, íleo o parálisis de cuerdas vocales. Además se produce una elevación de CK secundaria a la rabdomiolisis y que puede derivar en fracaso renal y coagulación intravascular diseminada que es la complicación más seria y conlleva una alta mortalidad. No todos los pacientes presentan todos los signos clínicos15. El diagnóstico se basa en las manifestaciones clínicas. Los desencadenantes pueden ser además de la disminución de la dosis de levodopa, el cambio de un agonista por otro, una infección, una elevación de la temperatura ambiental (golpe de calor) que produce deshidratación o modificaciones en la distribución de la levodopa por cambios en la alimentación (inicio de nutrición enteral). A veces consiste en un empeoramiento brusco sin que se haya suspendido el tratamiento antiparkinsoniano y sería la expresión de un superoff. En cuanto a la fisiopatología, se piensa que se produce una hipotransmisión dopaminérgica aguda en el hipotálamo, sistema nigroestriado, y sistema dopaminérgico mesocortical. Además existe una susceptibilidad individual16. En los estudios postmortem se ha encontrado una necrosis hipotalámica17.
El tratamiento consiste en su precoz reconocimiento y hay que administrar fluidos (2500-3000 ml de sueros/24 h) con electrolitos y glucosa, vitamina B1, medidas físicas y farmacológicas contra la hipertermia, tratar la infección si se diagnostica y reinstaurar el tratamiento antiparkinsoniano lo antes posible, por sonda nasogástrica si existen problemas de deglución (Tabla 4). También se han empleado bromocriptina, amantadina o apomorfina intravenosa. Si la rigidez es muy intensa puede ser necesario usar dantrolene a dosis de 80 mg/día por vía intravenosa16.
Psicosis aguda
Las manifestaciones clínicas de la psicosis en la EP consisten en alucinaciones que suelen ser visuales y estereotipadas y delirios que suelen ser de ruina, persecutorios o celotipias. Cuando las alucinaciones y delirios son intensos se acompañan de agitación, confusión y alteraciones del sueño, lo que altera la calidad de vida del paciente y de sus cuidadores. La psicosis está relacionada con el tratamiento antiparkinsoniano, pero no se debe sólo a la disfunción dopaminérgica sino que intervienen otras alteraciones neuroquímicas. Se ha propuesto que las alucinaciones precoces se deben a una hipersensibilidad de los receptores mesolimbocorticales y las que aparecen de forma tardía a mecanismos no puramente dopaminérgicos sino a degeneración de circuitos serotoninérgicos o colinérgicos18.
Cuando la psicosis es aguda se deben investigar factores desencadenantes como infecciones, alteraciones metabólicas o procesos cerebrales estructurales. Si los síntomas psicóticos son leves se debe tratar de simplificar el tratamiento antiparkinsoniano, reduciendo o suspendiendo fármacos añadidos a la levodopa: amantadina, selegilina, agonistas dopaminérgicos e inhibidores de la COMT19. Si a pesar de estas modificaciones los síntomas persisten hay que tratar de reducir la levodopa y si esto no fuera suficiente se debe administrar neurolépticos atípicos como clozapina, quetiapina, zisapridona o aripiprazol20. Los inhibidores de la colinesterasa han demostrado ser eficaces en los casos de EP con demencia21. También se ha propuesto el ondansetrón, antagonista 5HT3 como alternativa terapéutica a los neurolépticos22.
Urgencias en pacientes con neuroestimuladores cerebrales profundos
La estimulación cerebral profunda es un tratamiento consolidado como alternativa terapéutica en pacientes seleccionados con EP. Este tratamiento puede conllevar complicaciones que ocasionen consultas en urgencias que deben ser conocidas por el neurólogo general a pesar de que en estas circunstancias el paciente debe acudir a un centro especializado23.
Dentro de las complicaciones quirúrgicas diferidas se encuentran la trombosis venosa profunda y el tromboembolismo pulmonar como en cualquier otro procedimiento quirúrgico que conlleve encamamiento prolongado. Pueden aparecer infecciones locales así como serosas o acúmulos de líquido en las bolsas quirúrgicas donde se aloja el estimulador y complicaciones derivadas del material implantado como desplazamiento o rotura del electrodo, o rotura del cable de conexión por infecciones o necrosis en su trayecto subcutáneo. Puede ocurrir que se desconecte el generador por contacto fortuito con un campo magnético intenso produciendo un empeoramiento súbito de los síntomas parkinsonianos, lo que sucede también si se agotan las baterías del neuroestimulador que suelen durar unos cinco años. Tanto en estos casos como ante la sospecha de un efecto negativo inducido por un cambio reciente en los parámetros del neuroestimulador es necesario contar con neurólogos expertos en el manejo de su sistema de programación.
Complicaciones derivadas de la propia estimulación cerebral son la aparición de trastornos de movimiento de tipo coréico o balístico por estímulo del núcleo subtalámico. También pueden producirse trastornos psiquiátricos o cognitivos como abulia, apatía, anhedonia, euforia o hipomanía que se han relacionado con la estimulación de áreas mediales y ventrales del núcleo subtalámico. La estimulación de fibras vecinas de la cápsula interna puede producir síntomas pseudobulbares como disfagia, disartria y llanto inapropiado.
El neuroestimulador produce un artefacto eléctrico en los electrocardiogramas, electroencefalogramas y un artefacto por efecto metálico en las tomografías computerizadas. Estos efectos deben ser tenidos en cuenta cuando un paciente portador de un neuroestimulador consulta en urgencias por otros problemas médicos24.
Parkinsonismo agudo
La aparición de un parkinsonismo de forma aguda es muy infrecuente. Puede estar causado por agentes infecciosos como el Mycoplasma o por distintos fármacos como la anfotericina B1. Trastornos metabólicas como la mielinolisis central pontina o extrapontina pueden producir parkinsonismo agudo25. Esta entidad aparece como consecuencia de trastornos osmóticos como por el ejemplo en el contexto de la corrección de una hiponatremia secundaria a hiperemesis y se encuentran lesiones hiperintensas en el estriado que pueden ser reversibles así como el parkinsonismo. Existe una entidad hereditaria rara con ligamiento en una zona del cromosoma 19 denominada distonía-parkinsonismo de inicio rápido en el que los síntomas pueden aparecer de forma aguda o subaguda con parkinsonismo y distonía de musculatura orofacial26. La aparición de parkinsonismo por consumo accidental o por adicción de distintos tóxicos (Tabla 6) está documentada en la literatura27-29.

La encefalitis letárgica se produjo de forma epidémica a comienzos del siglo XX como una encefalitis con afectación de los ganglios basales produciendo parkinsonismo fundamentalmente y también trastornos psiquiátricos y del sueño. En las últimas décadas también se han descrito casos de esta encefalitis cuya fisiopatología no se conoce bien. No hay clara evidencia de agentes infecciosos que invadan el sistema nervioso aunque sí se encuentra linfocitosis en el líquido cefalorraquídeo y bandas oligoclonales. Puede haber cambios de señal en mesencéfalo y ganglios basales en la resonancia magnética craneal aunque también puede ser normal. En casos recientes se han usado esteroides con buena respuesta clínica y la recuperación ha sido completa en varios de ellos30.
Distonías agudas
Dentro de este grupo la causa más frecuente de consulta en urgencias son las distonías secundarias a fármacos. Suele verse en personas jóvenes a las primeras horas o días de tomar el tratamiento con fármacos antagonistas dopaminérgicos como los antipsicóticos (haloperidol) o antieméticos (cleboprida o metoclopramida), antidepresivos del grupo de los inhibidores selectivos de la recaptación de la serotonina o noradrenalina y tras el consumo de cocaína. Pueden aparecer también después del empleo de antipsicóticos atípicos31-33. Otros fármacos como la rivastigmina o el interferón alfa-2b pueden provocar ocasionalmente reacciones distónicas agudas34. Las manifestaciones clínicas consisten en distonía focal generalmente craneocervical, lo más frecuente es en forma de tortícolis, pero también puede ser en forma de trismos, profusión lingual, crisis oculogiras, distonía faríngea, laríngea, blefarospasmo e incluso axiales y en extremidades. Se presenta hasta en el 30-40% de los jóvenes psicóticos en los que se inicia el tratamiento con un antipsicótico clásico31. No parece tener relación con la dosis y tienen más riesgo los pacientes con SIDA y con inmunodeficiencias en los que se han descrito reacciones distónicas tras tratamientos con otros fármacos como las inmunoglobulinas intravenosas35. El tratamiento de ese efecto adverso consiste en fármacos anticolinérgicos por vía parenteral como el Akinetón® (biperideno) 5 mg iv o im o el Cogentin® (benztropina) 1-2 mg, la dosis puede repetirse si no mejoran los síntomas y es conveniente continuar el tratamiento unos días después por vía oral31.
Otro motivo de consulta urgente es la distonía laríngea aguda o síndrome de Gerhardt que consiste en una distonía esporádica idiopática en la que se produce un espasmo laríngeo por distonía en adducción de las cuerdas vocales durante la inspiración que puede llegar a provocar una obstrucción de la vía aérea. La manifestación clínica consiste en un estridor respiratorio que cede con el sueño. Es una entidad muy poco frecuente que se confirma por fibroscopia y el tratamiento consiste en la inyección de toxina botulínica en los músculos tiroaritenoideos31-36.
Los pacientes con atrofia multisistémica también pueden presentar un estridor laríngeo por una alteración en la abducción de las cuerdas vocales debida a espasmos distónicos de los músculos tiroaritenoideos (adductores) y debilidad de la musculatura abductora por atrofia neurógena del músculo cricoaritenoideo posterior. Es un síntoma grave que puede producir muerte súbita. Su tratamiento consiste en aplicar CPAP (ventilación pasiva no invasiva), toxina botulínica o traqueostomía31.
La tormenta distónica o estatus distónico supone una urgencia grave que puede aparecer en pacientes con distonías primarias o secundarias. La sintomatología consiste en la aparición de graves espasmos distónicos generalizados muy dolorosos que conllevan la aparición de hipertermia, rabdomiolisis con riesgo de fracaso renal agudo, anartria, insuficiencia respiratoria y disfagia con riesgo de neumonía por broncoaspiración. El estatus distónico comparte muchos aspectos del estatus epiléptico. Los factores precipitantes pueden ser los mismos: traumatismos, cirugía, infección, fiebre e introducción rápida o retirada brusca de algún fármaco. No hay datos establecidos sobre la estrategia óptima de tratamiento. Sólo en raras ocasiones los fármacos por vía oral (haloperidol, tetrabenazina o pimozide) son efectivos. El midazolam, benzodiacepina de acción rápida y vida media corta, se puede usar como fármaco de primera línea (30-100 µg/kg/hora), si falla puede usarse propofol (0,3-3,0 mg/kg/hora). Las medidas terapéuticas generales consisten en hidratación, evitar el fracaso renal agudo, analgesia, monitorización electrocardiográfica, vigilancia respiratoria e ingreso en UCI si es preciso. Como en el estatus epiléptico, en ocasiones es necesaria la anestesia general y el bloqueo neuromuscular con agentes curarizantes. En enfermedades degenerativas puede ser necesaria la infusión de baclofen intratecal o cirugía funcional37.
Urgencias pseudodistónicas
A veces una subluxación atlantoaxoidea o un absceso retrofaríngeo en niños puede presentarse como un tortícolis38 así como tumores medulares o de fosa posterior. También pueden incluirse en este apartado los trastornos psicógenos del movimiento o cuadros conversivos que suelen consistir en movimientos abigarrados y floridos que comienzan de forma aguda. Es importante considerar este diagnóstico para evitar exploraciones innecesarias aunque en ocasiones el diagnóstico diferencial puede ser difícil y es necesario mantener al paciente en observación1.
Corea-Balismo agudos
El balismo y la corea forman parte del mismo espectro de trastorno motor. El balismo proximal puede acompañarse de corea en la parte distal de la extremidad y puede transformarse en corea. El balismo o la corea de presentación aguda suelen deberse a lesiones estructurales del sistema nervioso generalmente de origen isquémico aunque en ocasiones otros procesos de diversa naturaleza (infecciones, enfermedades autoinmunes, hipoglucemia u otros trastornos metabólicos) pueden producir balismo agudo39. La corea puede ser la forma de presentación de una enfermedad paraneoplásica e iniciarse de forma aguda o subaguda pudiendo ser motivo de consulta en urgencias. Generalmente va a acompañado de otros síntomas de encefalopatía y en las pruebas de imagen se suelen encontrar alteraciones de señal en los ganglios basales. Se asocia a carcinoma de pulmón de células pequeñas y anticuerpos anti-Hu o anti-CV240,41.
Mioclonias
La mayor parte de las mioclonias o asterixis son una manifestación clínica de una encefalopatía metabólica por fallo hepático o renal. Las mioclonias pueden formar parte del síndrome serotoninérgico. La intoxicación por opiáceos puede acompañarse de mioclonias, así como el síndrome de abstinencia de estas drogas. El tratamiento con litio, antidepresivos tricíclicos, imipenem y cefuroxima pueden producir mioclonias de acción corticales. Lesiones focales cerebrales de distintas etiologías (infecciosa, vascular, etc.) pueden producir mioclonias focales1.
Trastornos del movimiento en el ictus
La presentación de un ictus con un trastorno del movimiento es infrecuente y sucede en el 1% de los casos. El trastorno del movimiento más frecuente secundario a un ictus es la hemicorea-hemibalismo seguido de la hemidistonía42. Las lesiones isquémicas que afectan al globo pálido o al mesencéfalo pueden producir parkinsonismo agudo43, los infartos del putamen pueden producir distonía y los infartos en el núcleo subtalámico o caudado suelen manifestarse con corea-balismo44. Los infartos talámicos pueden producir distintos trastornos del movimiento según los núcleos talámicos implicados. La lesión de los núcleos ventrales anteriores puede producir una distonía mioclónica (Fig. 1) y una mano talámica con postura distónica aislada con movimientos pseudoatetósicos por lesión en el núcleo ventral intermedio (VIM) y ventrales posteriores respectivamente y también temblor postural y de acción por lesión del VIM y alteración de la conexión cerebelotalámica45.
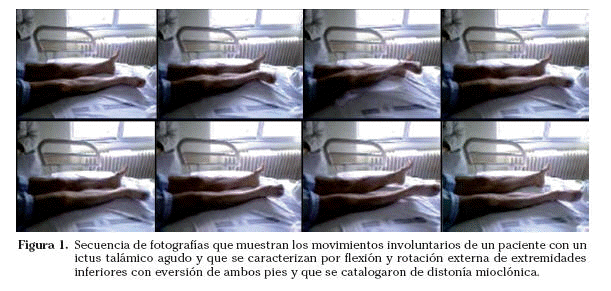
La etiología más habitual de los ictus, que se manifiestan con un trastorno del movimiento es la enfermedad de pequeño vaso aunque en el caso de los infartos talámicos hay que considerar la posibilidad de un origen cardioembólico46. La mayor parte de estos trastornos del movimiento que aparecen en la fase aguda son transitorios y desaparecen espontáneamente en los primeros días o semanas.
Se han descrito fenómenos motores involuntarios de presentación variada en las lesiones isquémicas del tronco cerebral que pueden consistir desde movimientos de pequeña amplitud semejantes a mioclonias hasta auténticas sacudidas de extremidades e incluso del tronco que suelen ser intermitentes y pueden desencadenarse por estímulos dolorosos47. Se desconoce con qué frecuencia aparecen pero sí se sabe que suelen presentarse en casos de lesiones extensas del tronco generalmente en relación a oclusión aguda del tronco basilar48. Su fisiopatología es incierta y se considera la isquemia del tracto corticoespinal como el mecanismo más probable. El reconocimiento de estos movimientos es importante ya que pueden hacer pensar en que se está produciendo una oclusión basilar y así establecer las medidas de diagnóstico y tratamiento con rapidez49.
En conclusión, en los pacientes que acuden a urgencias con un trastorno del movimiento de presentación aguda las manifestaciones clínicas pueden ser muy variadas así como su etiología, hay que atender los desórdenes autonómicos que pueden acompañar al cuadro clínico y establecer medidas terapéuticas que pueden en ocasiones requerir el ingreso en una unidad de cuidados intensivos (Fig. 2).

Bibliografía
1. Kipps CM, Fung VSC, Grattan-Smith P, de Moore GM, Morris JGL. Movement disorders emergencies. Movement disorders 2005; 20: 322-334. [ Links ]
2. Dressler D, Bebecke R. Diagnosis and management of acute movement disorders. J Neurol 2005; 252: 1299-1306. [ Links ]
3. Wood D, Webster E, Martinez D, Dargan P, Jones A. Case report: Survival after deliberate strychnine self-poisoning, with toxicokinetic data. Crit Care 2002; 6: 456-459. [ Links ]
4. Vetrugno R, Mascalchi M, Vella A, Della Nave R, Guerrini L, Vattimo A et al. Pontine hyperperfusion in sporadic hyperekplexia. J Neurol Neurosurg Psychiatry 2007; 78: 1001-1004. [ Links ]
5. Linazasoro G, Van Blercom N. Síndrome neuroléptico maligno y catatonía letal. Neurol Suppl 2007; 3: 2-6. [ Links ]
6. Kaplan PW, Birbeck G. Lithium-induced confusional status epilepticus or triphasic encephalopathy? Epilepsia 2006; 47: 2071-2074. [ Links ]
7. Admet P, Lestavet P, Krivosic-Horber R. Neuroleptic malignant syndrome. Br J Anaesth 2000; 83: 129-135. [ Links ]
8. Martínez-Castrillo JC, Mariscal Ortega A. Síndrome serotoninérgico e hipertermia maligna. Neurol Suppl 2007; 3: 7-14. [ Links ]
9. Mason PJ, Morris VA, Balcezak TJ. Serotonin syndrome: presentation of two cases and review of the literature. Medicine (Baltimore) 2000; 79: 201-209. [ Links ]
10. Boyer EW, Shannon M. The serotonin syndrome. N Engl J Med 2005; 352; 11: 1112-1120. [ Links ]
11. McCarthy TV, Healy JM, Heffron H. Localization of the malignant hyperthermia susceptibility locus to human chromosome 19q12-13.2. Nature 1990; 343: 562-564. [ Links ]
12. Abraham RB, Adnet P, Glauber V. Malignant hyperthermia. Post Grad Med J 1998; 74: 11-17. [ Links ]
13. García-Ruiz PJ, del valle M, Cabo I, García bermejo P. Síndrome de la persona rígida y otras causas de rigidez espinal aguda o subaguda. Neurol Suppl 2007; 3: 32-34. [ Links ]
14. Meinck HM, Thompson PD. Stiff man syndrome and related conditions. Mov Disord 2002; 17: 853-866. [ Links ]
15. Serrano-Dueñas M. Neuroleptic malignant syndrome-like, or (dopaminergic malignant syndrome) due to levodopa therapy withdrawal. Clinical features in 11 patients. Parkinsonism Relat Disord 2003; 9: 175-178. [ Links ]
16. Luquin piudo MR. Síndrome de acinesia aguda-hiperpirexia. Neurol Suppl 2007; 3:15-20. [ Links ]
17. Horn E, Lach B, Lapierre Y, Hrdina P. Hypothalamic pathology in neuroleptic malignant syndrome. J Neurol 1987; 235: 120-121. [ Links ]
18. Aarsland D, Larsen JP, Cummings JL, Laake K. Prevalence and clinical correlates of psychotic symptoms in Parkinson disease: a community-based study. Arch Neurol 1999; 56: 595-601. [ Links ]
19. Kulisevski J, Martínez-Ramírez S. Tratamiento de la psicosis en la enfermedad de Parkinson. Neurol Suppl 2007; 3: 21-26. [ Links ]
20. Gómez-Esteban JC, Zarranz JJ, Velasco F, Lezcano E, Lachen MC, Rouco I et al. Use of ziprasidone in parkinsonian patients with psychosis. Clin Neuropharmacol 2005; 28: 111-114. [ Links ]
21. Reading PJ, Luce AK, McKeith IG. Rivastigmine in the treatment of parkinsonian psychosis and cognitive impairment: preliminary findings from an open trial. Mov Disord 2001; 16: 1171-1174. [ Links ]
22. Zoldan J, Friedberg G, Livneh M, Melamed E. Psychosis in advanced Parkinsons disease: treatment with ondansetron, a 5-HT3 receptor antagonist. Neurology 1995; 45: 1305-1308. [ Links ]
23. Valldeoriola F, Pilleri M, Tolosa E, Molinuelo JL, Rumiá J, Ferrer E. Bilateral subthalamic stimulation monotherapy in advanced Parkinsons disease: long-term follow-up of patients. Mov Disord 2002; 17: 125-132. [ Links ]
24. Valldeoriola F. Urgencias en pacientes con neuroestimuladores implantados. Neurol Suppl 2007; 3: 40-46 [ Links ]
25. Sajith J, Ditchfield A, Katifi HA. Extrapontine myelinolisis presenting as acute parkinsonism. BMC Neurology 2006; 6:33 [ Links ]
26. Linazasoro G, Indakoetxea B, Ruiz J, Van Blercom N, Lasa A. Possible sporadic rapid-onset dystonia-parkinsonism. Mov Disord 2002; 17: 608-609. [ Links ]
27. Zandio B, Erro ME, Muñoz R, Mayor S, Ayuso T, Villanueva JA. Parkinsonismo agudo tras abstinencia alcohólica e ingesta oral del fungicida de uso agrario Mancozeb. Neurología 2007; 22: 704. [ Links ]
28. A de Bie RM, Gladstone RM, Strafella AP, Ko J-H, Lang AE. Manganese-induced parkinsonism associated with methcathinone (ephedrone) abuse. Arch Neurol 2007; 64: 886-889. [ Links ]
29. Mätzler W, Nägele T, Gasser T, Krüger R. Acute parkinsonism with corresponding lesions in the basal ganglia after heroin abuse. Neurology 2007; 68: 414. [ Links ]
30. Dale RC, Webster R, Gill D. Contemporary encephalitis lethargica presenting with agitated catatonia, stereotypy, and dystonia-parkinsonism. Mov Disord 2007; 22: 2281-2284. [ Links ]
31. Grandas F, Contreras A. Urgencias en distonía. Neurol Suppl 2007; 3: 35-39. [ Links ]
32. Corominas Sánchez M, Lleonart Bellfill R, Pujol Farriols R. Acute laryngeal dystonia due to clebopride simulating allergic reaction. Med Clin (Barc) 2007; 129:238-239. [ Links ]
33. Dingli K, Morgan R, Leen C. Tetanus versus acute dystonic reaction caused by metoclopramide. BMJ 2007; 334: 899-900. [ Links ]
34. Quarantini LC, Miranda-Scippa A, Parana R, Sampaio AS, Bressan RA. Acute dystonia after injection of pegylated interferon alpha-2b. Mov Disord 2007; 22: 747-748. [ Links ]
35. Papapetropoulos S, Friedman J, Blackstone C, Kleiner GI, Bowen BC, Singer C. A progressive, fatal dystonia-Parkinsonism syndrome in a patient with primary immunodeficiency receiving chronic IVIG therapy. Mov Disord 2007; 22: 1664-1666. [ Links ]
36. Papapetropoulos S, Papapetropoulos N, Singer C. Primary dystonia presenting as a respiratory emergency. Neurology 2007; 68: 388-389. [ Links ]
37. Mariotti P, Fasano A, Contarino MF, Della Marca G, Piastra M, Genovese O et al. Management of status dystonicus: our experience and review of the literature. Mov Disord 2007; 22: 963-968. Review [ Links ]
38. Simsek S, Yigitkanli K, Kazanci A, Belen D, Bavbek M. Medically treated paravertebral Brucella abscess presenting with acute torticollis: case report. Surg Neurol 2007 Feb; 67: 207-210. [ Links ]
39. Castro García A, Sesar Ignacio Á. Balismo. Neurol Suppl 2007; 3: 27-31. [ Links ]
40. Tani T, Piao Y-S, Mori S, Ishihara N, Tanaka K, Wakabayashi K et al. Chorea resulting from paraneoplastic striatal encephalitis. J Neurol Neurosurg Psychiatry 2000; 69: 512-515. [ Links ]
41. Muehlschlegel S, Okun MS, Foote KD, Coco D, Yachnis AT, Fernandez HH. Paraneoplastic chorea with leukoencephalopathy presenting with obsessive-compulsive and behavioral disorder. Mov Disord 2005; 20: 1523-1527. [ Links ]
42. Ghika-Schmid F, Ghika J, Regli F, Bogousslavsky J. Hyperkinetic movement disorders during and after acute stroke: the Lausanne Stroke Registry. J Neurol Sci 1997; 146: 109-116. [ Links ]
43. Padilla Parrado F, Campos Arillo VM, Martínez del Valle Torres MD, Ortega Lozano SJ. Hemiparkinsonismo secundario a hemorragia mesencefálica. Neurología 2007; 22: 480-483. [ Links ]
44. Krauss JK, Pohle T, Borremans JJ. Hemichorea and hemiballism associated with contralateral hemiparesis and ipsilateral basal ganglia lesions. Mov Disord 1999; 14: 497-501. [ Links ]
45. Lehéricy S, Grand S, Pollak P, Poupon F, Le Blas JF, Limousin P et al. Clinical characteristics and topography of lesions in movement disorders due to thalamic lesions. Neurology 2001; 57: 1055-1066. [ Links ]
46. Ajuria I, Erro ME, Ureña M, Ciriza M, Gómez ML, Gállego J. Infarto talámico anterior secundario a lipoma auricular derecho y foramen oval permeable. Neurología 2007; 22: 790. [ Links ]
47. Saposnik J, Caplan LR. Convulsive-like movements in brainstem stroke. Arch Neurol 2001; 58: 654-657. [ Links ]
48. Myung SL, Seung HO, Kiung RL. Transient repetitive movements of the limbs in patients with acute basilar artery infarction. Neurology 2002; 59: 1116-1119. [ Links ]
49. Herrera M, Erro ME, Aymerich N, Gállego J. Movimientos involuntarios en lesiones isquémicas del tronco cerebral. Neurología 2007; 22: 475-479. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
María Elena Erro Aguirre
Servicio de Neurología
Hospital de Navarra
Irunlarrea, 3
31008 Pamplona
Tfno. 848422292
Fax: 848422303
E-mail: elena.erro.aguirre@cfnavarra.es

