










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versión impresa ISSN 1137-6627
Anales Sis San Navarra vol.31 supl.2 Pamplona 2008
Genetically based diseases
D. González-Lamuño, M. García Fuentes
Servicio de Pediatría. Hospital Universitario Marqués de Valdecilla-Universidad de Cantabria. Santander.
Dirección para correspondencia
RESUMEN
La genética constituye uno de los mayores avances científicos del siglo XX, que comienza con el redescubrimiento de las leyes de Mendel y termina con la elaboración del primer borrador de la secuencia completa del genoma humano. La genética utiliza diferentes estrategias de investigación, como los estudios de gemelos y de adopción, que investigan la influencia de los factores genéticos y ambientales, y las estrategias para identificar genes específicos (genética molecular). Además del importante grado de discapacidad que generan, el impacto social de las enfermedades hereditarias es enorme, por su carácter potencialmente recurrente en una misma familia y por el elevado coste socio-sanitario derivado de la enorme carga de cuidados que requiere. El diagnóstico de las enfermedades hereditarias presenta características diferenciadoras muy significativas ya que el resultado de un diagnóstico genético tiene no sólo efectos sobre el paciente sino también sobre todos los individuos emparentados. Por tanto, la unidad de estudio en el diagnóstico genético es la familia y todo proceso de diagnóstico implica una investigación familiar. También conviene tener en cuenta que los protocolos de diagnóstico se desarrollan de forma paralela a la investigación básica y generalmente están poco estandarizados. Los resultados obtenidos en los estudios genéticos y el tipo de información que se facilita al paciente y a su familia deben ser matizados dentro del proceso del consejo genético.
Palabras clave. Enfermedades raras. Genética molecular. Bases de datos genéticos. Estudios genéticos. Investigación genética. Consejo genético.
ABSTRACT
Genetics is one of the greatest scientific advances of the XX century, which begins with the rediscovery of Mendels laws and culminates in the elaboration of the first draft of the complete sequence of the human genome. Genetics employs different research strategies, such as the study of twins and adoption, investigating the influence of genetic and environmental factors, and strategies for identifying specific genes (molecular genetics). Besides the significant degree of disability they generate, the social impact of hereditary diseases is enormous, due to their potentially recurrent character in the same family and the high socio-health cost deriving from the enormous care burden they require. The diagnosis of hereditary diseases presents very significant differentiating characteristics since the result of a genetic diagnosis has effects not only on the patient but also on related individuals. Thus the unit of study in genetic diagnosis is the family and the whole process of diagnosis involves family research. It is also useful to bear in mind that the protocols of diagnosis are developed in parallel with the basic research and in general are hardly standardised. The results obtained in genetic studies and the type of information provided to the patient and his family must be qualified within the process of genetic counselling.
Introducción
Las enfermedades de base genética constituyen un grupo de patologías muy importante, no sólo por su incidencia relativamente elevada, alrededor de un 1 por ciento de los bebés nacen con algún tipo de anormalidad genética, sino por el tipo de problemas que producen. Desde el punto de vista clínico estas enfermedades se caracterizan por comprometer la calidad de vida de los afectos, causando una grave discapacidad intelectual o física. Así mismo, es frecuente que estas enfermedades tengan un carácter progresivo y condicionen una mortalidad precoz. En determinadas enfermedades genéticas que causan un deterioro progresivo e inexorable, un diagnóstico puede además suponer virtualmente una sentencia de muerte precoz1.
Un elevado porcentaje de las denominadas enfermedades raras tienen un origen genético, de ahí la gran importancia que tiene para este conjunto de enfermedades la genética como ciencia y como aproximación diagnóstica, para entender aspectos fundamentales de estas enfermedades y para el adecuado manejo clínico de las mismas. Este conocimiento es también el fundamento de los servicios clínicos que se ofrecen a los pacientes y familiares, desde el diagnóstico y asesoramiento genético hasta la investigación de las bases moleculares y celulares que explican los mecanismos de producción y la fisiopatología de las enfermedades, abriendo un campo de enormes posibilidades para definir dianas moleculares que sean la base de nuevas terapias2.
Hablar de genética en las enfermedades raras es referirse al conjunto amplio de enfermedades monogénicas, síndromes cromosómicos y malformaciones congénitas. Las enfermedades monogénicas son trastornos debidos a mutaciones que pueden afectar bien a alguno de los aproximadamente 25.000 genes del genoma nuclear que codifican proteínas y se transmiten según las leyes de la herencia de Mendel (de ahí que también se conozcan como enfermedades mendelianas), o en el pequeño genoma ubicado en la matriz de las mitocondrias con un patrón de transmisión específico conocido como herencia mitocondrial. Las aberraciones cromosómicas son el sustrato etiopatogénico de síndromes clásicos debidos a alteraciones numéricas de los cromosomas (síndrome de Down, síndrome de Turner, síndrome de Klinefelter), deleciones parciales, ejemplos de las cuales son el síndrome del maullido del gato, el síndrome de Williams y el síndrome de microdeleción 22q11, o a alteraciones estructurales de los cromosomas1.
La enfermedad de Huntington y la fenilcetonuria son respectivamente ejemplos de enfermedades hereditarias dominantes y recesivas que siguen las reglas básicas de la herencia descrita por Mendel hace más de un siglo. Un gen puede existir en dos o más formas diferentes (alelos) y uno de los alelos puede dominar la expresión del otro. Los dos alelos, uno de cada progenitor, se separan (segregan) durante la formación de los gametos. Esta circunstancia fundamenta la primera ley de Mendel, la ley de la segregación. La ley explica muchas características de la herencia: el hecho de que el 50% de los descendientes de una persona con la enfermedad de Huntington desarrollan finalmente la enfermedad; la persistencia de que este gen letal persista en la población; el hecho de que los niños con fenilcetonuria no tengan, por lo general, padres con esa enfermedad y por qué la fenilcetonuria aparece con mayor probabilidad cuando los padres están emparentados.
La segunda ley de Mendel es la ley de la combinación independiente: la herencia de un gen no se ve afectada por la herencia de otro gen. Sin embargo, los genes que están muy próximos en el mismo cromosoma, ligados, pueden transmitirse juntos incumpliendo la ley mendeliana de la combinación independiente. Tales excepciones hacen posible la elaboración de mapas utilizando el análisis del ligamiento y de esta forma ya se han identificado los genes responsables de la enfermedad de Huntington y de la fenilcetonuria.
Las leyes de la herencia de Mendel no explican todos los fenómenos genéticos. Por ejemplo, los genes situados en el cromosoma X, tales como el gen que determina la ceguera para los colores, necesitan una extensión de las leyes mendelianas. Otras excepciones de las leyes incluyen mutaciones nuevas, cambios en los cromosomas, como la no disyunción que produce el síndrome Down, la expansión de tripletes repetidos que originan la enfermedad de Huntington y la deficiencia mental por X frágil y el fenómeno de la impronta genómica.
La mayoría de los aspectos y trastornos psicológicos de los individuos muestran patrones de herencia más complejos que las alteraciones monogénicas como la enfermedad de Huntington, la fenilcetonuria o la ceguera para los colores. Trastornos complejos como la esquizofrenia, o caracteres con distribución continua como la habilidad cognitiva general, probablemente se hallen influidos por muchos genes así como por factores ambientales. La genética cuantitativa aplica las reglas de la herencia monogénica a los sistemas poligénicos. El fundamento de esta disciplina es que los caracteres complejos pueden estar influidos por muchos genes, pero cada gen se transmite según las leyes de Mendel. Los métodos que se emplean, especialmente los estudios de gemelos y los hijos adoptados, permiten detectar la influencia genética que reciben estos caracteres complejos3.
Muchas de estas enfermedades afectan al sistema nervioso con lo que adquieren una importancia singular, porque atañen a aquello que caracteriza de modo más específico a los seres humanos: el comportamiento cognitivo, la memoria, la conducta emocional, etc. Como corresponde a un órgano tan complejo, existen múltiples patologías genéticas capaces de producir una disfunción del sistema nervioso, cuya repercusión sobre el desarrollo del individuo en ocasiones es poco predecible, ya que puede ser modificada por cambios en el flujo sanguíneo cerebral, respuestas inmunitarias, infecciones, agresiones por factores ambientales, etc.
Además del problema individual de estas enfermedades debido al importante grado de discapacidad que generan, el impacto social de las enfermedades hereditarias es enorme por su carácter potencialmente recurrente en una misma familia y por su elevado coste socio-sanitario. En términos de utilización de recursos sanitarios, sin contar el número de consultas, se calcula que la patología genética es directamente responsables de uno de cada diez ingresos hospitalarios infantiles e indirectamente responsables de la mitad de dichos ingresos. Adicionalmente hay que tener en cuenta la enorme carga de cuidados que representa para otros miembros de la familia y para la sociedad en general.
Sin embargo, no podemos considerar que todas las enfermedades genéticas carecen de tratamientos específicos. Quizá el mejor ejemplo sean algunos graves trastornos metabólicos como la fenilcetonuriaa, cuya prevención es sencilla: los niños diagnosticados de fenilcetonuria que desde el nacimiento llevan una dieta baja en fenilalanina se desarrollan normalmente. En esta enfermedad por lo tanto, la nutrición por sí misma determina el que se produzca un desarrollo cerebral normal o una discapacidad profunda. Queda claro que después del nacimiento conviene saber lo antes posible cuál es la situación respecto a la fenilcetonuria, y lógicamente de muchas otras enfermedades cuyo tratamiento precoz será capaz de modificar su curso4.
Las mejoras en la comprensión y el diagnóstico de muchas de estas enfermedades se deben a los avances en el conocimiento de sus bases moleculares y a la capacidad tecnológica de aislar y clonar genes relacionados directamente con ellas. Tras el diagnóstico molecular emerge la posibilidad de prevenir el nacimiento de niños con enfermedades genéticas y la teórica posibilidad de alterar genéticamente las células de los individuos enfermos para mejorar su pronóstico. La realidad es que a pesar de los avances que se han producido en el diagnóstico y la comprensión patogénica de muchas enfermedades genéticas, en la mayor parte de los casos aún estamos lejos de disponer de soluciones terapéuticas definitivas. En un futuro es previsible que muchos individuos se beneficien de algunos aspectos de la investigación basada en la terapia génica o de células germinales, pero es poco probable que los avances en estas técnicas vayan a suponer algún tipo de mejora en el tratamiento de aquellas personas con una enfermedad genética que en la actualidad presentan discapacidad intelectual o física. A corto plazo, es previsible que únicamente se beneficien de este tipo de tratamientos los pacientes afectos de enzimopatías o determinados errores congénitos del metabolismo5. Existen sin embargo, muchos caminos de aproximación al estudio de las enfermedades de base genética que sí pueden suponer un beneficio inmediato para los pacientes o sus familiares y que pasan por un mejor conocimiento de los problemas últimos que condicionan la discapacidad. Diseccionando desde el punto de vista genético los síndromes debidos a extensos defectos genéticos, o valorando las intervenciones que se llevan a cabo en estos pacientes desde el punto de vista de respuesta biológica, podemos conseguir cambios significativos en el pronóstico de las enfermedades de base genética. Deben tenerse muy en cuenta además todos los aspectos de prevención y de consejo genético, así como la individualización de tratamientos médicos de acuerdo a las características propias de los individuos.
De forma paralela a estos avances en biomedicina, durante estos años hemos experimentado un enorme desarrollo en las técnicas de comunicación y en el acceso a la información. Grandes proyectos impulsores del desarrollo de la era molecular, como es el proyecto Genoma Humano, han generado una ingente cantidad de información que llega rápidamente tanto a profesionales como a la población general, abriéndose de este modo nuevas esperanzas terapéuticas para las enfermedades de base genética, al tiempo que se generan una serie de conflictos éticos y de adecuación de los recursos sanitarios disponibles6.
Peculiaridades del diagnóstico genético
Si bien el objetivo final del diagnóstico genético, como el de todo diagnóstico médico, esencialmente consisten en identificar la causa de una enfermedad, el diagnóstico de las enfermedades hereditarias presenta características diferenciadoras muy significativas. A este respecto es de destacar que el resultado de un diagnóstico genético tiene no sólo efectos sobre el paciente sino también sobre todos los individuos emparentados con él mismo. Por lo tanto, la unidad de estudio en el diagnóstico genético es la familia y todo proceso de diagnóstico implica un estudio familiar.
También conviene tener en cuenta que los protocolos de diagnóstico se desarrollan de forma paralela a la investigación básica y generalmente están poco estandarizados. Los laboratorios de diagnóstico genético tienen por lo general un carácter híbrido al desarrollar actividades de investigación básica conjuntamente con la aplicación de dicha investigación al ámbito clínico. Frecuentemente el desarrollo de una estrategia diagnóstica es en sí misma una línea de investigación básica en la cual está implicada la caracterización de un gen y el espectro de mutaciones que causan la patología en estudio.
Otro aspecto que singulariza el diagnóstico genético es que con frecuencia debe dar respuesta a necesidades clínicas urgentes con una dimensión ético-social importante. En aquellos casos en que el diagnóstico se realiza durante los períodos prenatal y neonatal se requiere una respuesta rápida y precisa. En el primer caso para dar a los padres una información completa y fiable que les permita la toma de decisiones durante las primeras semanas de gestación y en el segundo caso para poder dar una rápida respuesta terapéutica en relación al recién nacido.
Por último, debemos tener en cuenta que no siempre la conclusión que se obtiene en el diagnóstico genético es determinante. Salvo en el caso de la detección directa de la mutación responsable de la patología, el resto de conclusiones diagnósticas tienen un mayor o menor componente probabilístico. Por ello, los resultados obtenidos y el tipo de información que se facilita al paciente y a su familia deben ser matizados en este sentido1.
Accesibilidad de las pruebas genéticas
Actualmente pueden diagnosticarse muchas enfermedades raras mediante análisis biológicos, que suele ser genético. Estos análisis son un elemento importante en la atención a estos pacientes, ya que permiten un diagnóstico precoz, y eventualmente un cribado en cascada familiar o una prueba prenatal. Dado el gran número de pruebas y la necesidad de diseñar y validar un conjunto específico de análisis de diagnóstico para cada una de ellas, ningún centro puede ser autosuficiente en este terreno. Es preciso intercambiar material de pacientes y pruebas más allá de las fronteras nacionales, consiguiéndose de esta forma paliar una carencia significativa en materia de disponibilidad de análisis para enfermedades raras. Es necesario facilitar este intercambio de muestras biológicas y resultados mediante normas y procedimientos claros, transparentes y consensuados a escala de la Unión Europea. Se requiere reducir las diferencias reglamentarias entre países en materia de confidencialidad, reembolso, transporte, almacenamiento de muestras y certificación de laboratorios.
Conviene instar a los laboratorios a que participen en pruebas de aptitud, prestando especial atención a sus resultados en materia de notificación. Hay que velar por que exista consejo genético previo y ulterior a los análisis. Esto requiere un apoyo apropiado (según el número de análisis por año) a los laboratorios de referencia. En los dos últimos años, instituciones interesadas en el marco de la Comisión Europea, el Consejo de Europa y en particular la OCDE, han trabajado a favor de una política de refuerzo de la calidad de los laboratorios2.
Implicaciones de los resultados de las pruebas genéticas
El conocimiento genético en ocasiones puede crear dilemas éticos. La amniocentesis, por ejemplo, es capaz de detectar otros problemas además de la trisomía del 21 que pueden exceder la información solicitada por el obstetra o la paciente, evitándose esta circunstancia con la utilización de técnicas de hibridación in situ que sólo revelan el número de cromosomas 21 presentes. Al ser cada vez más sofisticadas, los resultados de determinadas pruebas genéticas tienen consecuencias que llegan más allá de las razones por las que se solicitaron, afectando a veces a las vidas de individuos que no se las han hecho. Donde esto se hace más evidente es en las familias con historiales de alguna enfermedad hereditaria debida a defectos en un único gen. En estos casos, el diagnóstico no lo hace un citogenetista sino un biólogo molecular, que analiza fragmentos específicos de ADN en lugar de enormes cromosomas. El ADN se extrae de una muestra de tejido, obtenida en el caso del feto por amniocentesis, o, en el caso de un niño o de un adulto, mediante una muestra de sangre o de células de la mucosa oral. Hoy en día, la prueba consiste normalmente en amplificar con PCR la región crítica de la muestra de ADN –el gen sospechoso–, que posteriormente se secuencia para determinar si tiene o no la mutación. Los resultados de un individuo pueden informar de la situación genética de sus familiares. Lo que yo puedo llegar a conocer sobre mis genes implica a mis parientes biológicos, les preocupe a ellos saberlo o no7.
Para ilustrar la diferencia entre el diagnóstico genético y cualquier diagnóstico de otro tipo podemos tomar como ejemplo la indicación para realizar una prueba para detectar la enfermedad de Huntington, enfermedad neurológica fatal muy discapacitante una vez comienzan a aparecer los síntomas, a partir de la cuarta o quinta década de la vida. Un varón joven cuyo abuelo paterno haya muerto de esta enfermedad, puede solicitar que le hagan una prueba para detectar la enfermedad, mientras que su padre, pudo haber decidido no hacerse la prueba, prefiriendo vivir con esa incertidumbre del 50 por ciento antes que conocer su situación con certeza. Como el Huntington se manifiesta relativamente tarde en la vida, es posible que el padre sea portador de la mutación aunque todavía los síntomas no hayan aparecido. El joven sabe que su probabilidad de tener la mutación –y por lo tanto de desarrollar la enfermedad en el futuro– es a priori de una entre cuatro, pero puede querer conocer con seguridad su situación genética para esta enfermedad. El problema que se plantea es el siguiente: si él realmente tiene la mutación, inevitablemente tiene que haberla recibido de su padre, lo que significa que el padre también desarrollará la enfermedad. La pregunta del hijo sobre su situación genética contraviene directamente el deseo del padre de evitar conocerla. Su deseo de saber entra en conflicto con el deseo de su padre que evita conocer lo que puede ser una sentencia de muerte devastadora8.
A veces las implicaciones pueden no tener importancia para las generaciones actuales, pero sí para las futuras. El síndrome del cromosoma X frágil es una de las causas de deficiencia mental más comúnb. Además de un cociente intelectual bajo, los síntomas clásicos incluyen un fenotipo distintivo y un temperamento hiperactivo, ocasionalmente irritable. Se trata de una enfermedad ligada al sexo (el gen responsable se encuentra en el cromosoma X), pero la padecen tanto las mujeres como los hombres. Es evidente que una copia normal del gen no es suficiente para que el efecto del gen mutado sea inapreciable. Con todo, las mujeres tienden a padecer menos síntomas y la incidencia de la enfermedad entre ellas es menor de 1 entre 5.000 comparada con 1 entre 2.500 en los varones. El síndrome del cromosoma X frágil está causado por una mutación que consiste en la presencia de tripletes CGG en una determinada región del ADN, que se repite una y otra vez. Los individuos normales tienen unas treinta repeticiones, mientras que los portadores del síndrome del cromosoma X frágil tienen por lo menos cincuenta y a veces hasta noventa. Por razones no suficientemente comprendidas, el número de repeticiones tiende a aumentar en cada generación y, una vez que hay alrededor de doscientos treinta tripletes CGG, el gen no puede producir ARN mensajero y por lo tanto deja de funcionar. La enfermedad toma su nombre de una debilidad estructural visible en el cromosoma X mediante el cariotipo, causada por todas esas repeticiones. A la vez que el número de repeticiones va en aumento de una generación a la siguiente, la gravedad de la enfermedad también aumenta. Los últimos descendientes de un pedigrí con síndrome del cromosoma X frágil tienen un número mayor de repeticiones, y la enfermedad aparece en ellos en una forma más grave que en aquellos de quienes la han heredado. Los genetistas pueden entonces identificar a individuos que son portadores de una premutación, lo que significaría tener pocas repeticiones para causar problemas de momento, pero sí las suficientes como para desembocar en un síndrome del cromosoma X frágil en generaciones posteriores, teniendo en cuenta el aumento que se producirá en la generación siguiente. No sabemos todavía con exactitud la función de la proteína codificada por el gen afectado, pero parece que se une a las moléculas del ARN mensajero en las sinapsis o conexiones entre las células nerviosas9.
Cribado poblacional
Muchas mujeres, especialmente aquellas con hijos afectos de alguna enfermedad genética pueden hacerse la pregunta de por qué cuando estaban embarazadas no hicieron las pruebas para detectar determinadas enfermedades genéticas discapacitantes. La respuesta a esta cuestión es que en el momento actual vivimos los inicios de una revolución genética que ha transformado la tecnología médica, pero se mantiene una enorme distancia entre el progreso científico y la atención a los pacientes. En realidad, lo cierto es que a muchas mujeres sencillamente no se les informa de sus opciones, y que las pruebas de las que disponemos en la actualidad están notablemente infrautilizadas. En el mejor de los casos, las pruebas para detectar algunas enfermedades genéticas conocidas se realizan normalmente sólo en aquellas familias que ya tienen un miembro afectado. El razonamiento para establecer dicha limitación es que estos trastornos son raros y las pruebas muy costosas, argumento de carácter social que puede ser discutible en muchos casos. En otras ocasiones, las dificultades técnicas de la prueba tienen que ver con la variabilidad del defecto subyacente, habiéndose descrito para muchas enfermedades miles de mutaciones diferentes responsables, lo cual tiene importantes repercusiones en el diseño de un programa de cribado de la población. En estas situaciones, ante un test negativo difícilmente se podría afirmar con total seguridad que una pareja no corre el riesgo de engendrar hijos con una enfermedad concreta.
Otras reticencias hacia el examen generalizado se basan en razones menos materiales. Hay quienes consideran la criba como la admisión del fracaso, como una solución errónea. Los grupos de apoyo tratan de conseguir que las personas que padecen una enfermedad genética se sientan parte de la comunidad y sean valoradas por la sociedad. ¿Cómo se puede reconciliar ese objetivo con el examen generalizado que en definitiva lo que promueve es el aborto de los fetos afectados?
A pesar de su indiscutible utilidad, en el momento actual los exámenes genéticos suponen una fuente de polémica de carácter tanto individual como social. Aunque idealmente podría indicarse la realización de pruebas genéticas indiscriminadas para tratar de detectar precozmente un gran número de enfermedades de base genética, tanto monogénicas como poligénicas, realmente en el momento actual carecemos de mecanismos adecuados para manejar los resultados de estas pruebas.
El incesante incremento del esfuerzo por facilitar una atención sanitaria al alcance de toda la población debería, por sí solo, constituir un argumento sólido para que todas las madres tuviesen la oportunidad de hacerse pruebas genéticas prenatales, siempre y cuando dispongan de la información adecuada. El destino de los embarazos con defectos genéticos graves deberían decidirlo las propias embarazadas. Idealmente, una sociedad libre no debería exigir a una mujer que impida el desarrollo de un feto con una enfermedad genética, ni tampoco obligarla a que lo lleve a término; ni todas las mujeres son capaces de criar a un niño discapacitado, ni todas lo son de interrumpir un embarazo en función de la previsible calidad de vida del niño. Cualquiera que sea la decisión individual, el hecho sigue siendo que la criba reduce la aflicción constituyendo un bien social teórico, pero realmente no se dispone de recursos suficientes para manejar adecuadamente toda la información de posibilidades preventivas, terapéuticas o reproductivas.
A pesar del frustrante rechazo a aprovechar las ventajas de un examen genético a gran escala y la corta historia de esa práctica, existen numerosos estudios piloto realizados en poblaciones definidas, cuyo grado de repercusión socio-sanitaria es variable, así como la polémica que generan. En este sentido, algunos programas de diagnóstico de determinadas enfermedades genéticas en poblaciones de alto riesgo han sido considerados un éxito incluso en sociedades muy conservadoras. La enfermedad de TaySachs o idiocia amaurótica familiar, es una dolencia terrible, cien veces más habitual entre los judíos asquenazíes que en los grupos no judíos, que probablemente se deba a la presencia casual de una mutación desgraciada en las pequeñas poblaciones fundadoras. Los bebés con TaySachs nacen con apariencia normal, pero su desarrollo se va haciendo gradualmente más lento y pronto empiezan a quedarse ciegos. A los dos años de edad, sufren crisis neurológicas, hipotonía y un deterioro progresivo hasta que fallecen a los cuatro años ciegos y con una clínica de parálisis cerebral.
A partir de 1985, año en que se codificó el gen y se identificó la mutación causante de la enfermedad de TaySachs, se dispuso de la capacidad para desarrollar un test prenatal infalible en una población bien definida: unas condiciones hechas a medida para la ejecución con éxito de un programa de detección. Pero el examen prenatal ofrece un único remedio en caso de un diagnóstico positivo: el aborto, que por lo menos entre el segmento más ortodoxo de los asquenazíes está prohibido. Afortunadamente, también es posible examinar a los padres potenciales, y de esta manera se pudo encontrar una solución moralmente aceptable consistente en un programa dirigido a las parejas. En 1985, se estableció un programa para realizar la prueba para detectar la enfermedad de TaySachs en la comunidad judía ortodoxa local, animando a los jóvenes a que la realicen durante la época del instituto y de la universidad, porque durante esos años se pueden hacer la prueba gratuitamente. Un aspecto poco común de este programa es su extrema confidencialidad, ya que ni siquiera a los que se han hecho la prueba se les informa de si son portadores. A cada uno se le asigna un código y más tarde, cuando dos personas piensan contraer matrimonio, se cruzan los códigos y únicamente si los dos son portadores se revela la situación de ambos junto con la posibilidad de consejo. Esta revelación basada sólo en la necesidad intenta evitar la estigmatización de los portadores a la vez que se procura combatir la amenaza de la enfermedad de TaySachs10.
Gracias a este programa se ha reducido la incidencia de la enfermedad y debería ser considerado como un éxito incuestionable, pero hay quienes dentro de la comunidad judía le encuentran faltas al programa, considerándola intimidatoria e incluso eugenésica. Sin embargo, el programa disfruta de un gran apoyo por parte de la comunidad a la que sirve, demostrando que un programa de diagnóstico puede ser a la vez efectivo y culturalmente sensible, y que funciona incluso en una situación en la que en principio parecería que las tradiciones sociales y los preceptos religiosos podrían ir en contra de las pruebas genéticas.
Detección neonatal: Relación coste / Beneficio
Las nuevas tecnologías obligan a plantearse la posibilidad de ampliar los programas de cribado neonatal en curso para tratar de identificar situaciones clínicas que pudieran modificarse mediante tratamientos específicos, y desarrollar líneas de investigación en la prevención de enfermedades genéticas que se manifiestan clínicamente con retrasos de desarrollo, formas de autismo, epilepsia intratable, muerte súbita, o determinadas enfermedades psiquiátricas.
La capacidad tecnológica para emprender la detección sistemática neonatal de más enfermedades plantea una serie de interrogantes. Un programa de este tipo siempre genera algún tipo de beneficio para los pacientes y las familias afectadas y contribuye decididamente a aumentar los conocimientos de la enfermedad y de su epidemiología. Pero es necesario que la relación coste / beneficio sanitario sea muy claramente positiva, ya que desde un punto de vista pediátrico y social siempre estará latente la cuestión de si los fondos destinados a enfermedades genéticas relativamente raras no tendrían un mayor impacto sobre la salud si se dirigieran a problemas más frecuentes. Existe asimismo el problema de la detección de portadores de defectos genéticos de enfermedades recesivas, y que por tanto no van desarrollar ninguna enfermedad, en los que la identificación del estado de portador va a generar una serie de problemas psicológicos que tienen un costo social y sobre los que es obligado intervenir para evitar una iatrogenia debida al programa de detección masivo.
Otros importantes factores a tener en cuenta son las direcciones y objetivos en materia de salud que plantee la sociedad y el impacto psicosocial, ya que para algunas enfermedades han sido las propias asociaciones de padres de niños afectados las que han impulsado programas cuya indicación objetiva es muy discutible. En cualquier caso, y una vez tomada la decisión, es necesario elaborar modelos que contemplen globalmente las diferentes formas de lucha contra enfermedades implicadas, en función de la estructura de salud en la cual se han de integrar.
Estrategias en el diagnóstico molecular
El análisis de numerosas patologías genéticas es hoy técnicamente posible, debiéndose insistir en que el punto de partida de toda estrategia de diagnóstico molecular es el razonamiento clínico. Todo proceso de diagnóstico genético conlleva una precisa caracterización clínica del caso índice o propósitus, que constituye la clave de los resultados que se pueden esperar de un diagnóstico molecular. Exceptuando algunas cromosomopatías, en la mayoría de las enfermedades de base genética es poco probable que el diagnóstico molecular vaya a ser de utilidad si no se realiza previamente una adecuada orientación clínica. En muchas ocasiones, el diagnóstico genético es un elemento más de un diagnóstico diferencial en el que se pretende confirmar o contrastar, frente a distintas opciones, la causa de una determinada patología previamente caracterizada clínicamente. En este sentido, es imprescindible la colaboración entre el personal clínico y el genetista en la elaboración del historial médico del paciente. Para el diagnóstico, frecuentemente necesitamos determinadas exploraciones complementarias. Éstas incluyen, cariotipo en sangre y otros tejidos, análisis bioquímicos de sangre y orina, cultivos celulares, estudios radiológicos, estudios moleculares, etc.
Son muchas las estrategias para el diagnóstico genético, y en ocasiones es posible obtener resultados similares con aproximaciones diferentes. Dichas estrategias podrían ser clasificadas como directas o indirectas. Las estrategias directas son aquellas en las que realizamos el diagnóstico identificando las diferentes mutaciones del gen en cuestión. Aunque a priori parece una estrategia sencilla, no lo es tanto si tenemos en cuenta que el número de enfermedades producidas por más de un tipo de mutación en un mismo o diferentes genes, supera a aquellas que son consecuencia de una única mutación. Las estrategias de diagnóstico indirecto no implican el conocimiento del gen responsable, y se fundamentan en el estudio de la herencia conjunta de marcadores anónimos y el locus de la enfermedad estudiada. Para este fin, es necesario que el marcador utilizado presente un fuerte ligamento con el locus de interés, además de otras características que lo harán más o menos adecuado para el diagnóstico. Esta estrategia es útil a la hora de identificar rasgos heredados en una familia con una enfermedad genética determinada11.
En cuanto a las técnicas a realizar, si se sospecha que la enfermedad está asociada a una alteración cromosómica conocida, deberemos iniciar la investigación con un estudio citogenético. Un número importante de patologías genéticas están causadas por alteraciones cromosómicas, sobre todo aquellas que se manifiestan de forma sindrómica. Si nuestra patología pertenece a este grupo, la estrategia diagnóstica más adecuada será la caracterización cromosómica del propósitus y de su familia. Dicha caracterización consistirá básicamente en la elaboración del cariotipo para el estudio de posibles alteraciones estructurales o numéricas. La realización de un cariotipo de bandeo cromosómico estándar (300-500 bandas) o de alta resolución (más de 800 bandas), va a depender, respectivamente, de la sospecha de anomalías numéricas o estructurales y del tamaño esperado de la alteración. Ante un niño con defectos congénitos, hay que realizar siempre un cariotipo con bandas de alta resolución.
Otra técnica de la que se dispone actualmente es la hibridación in situ (FISH), que combina el estudio citogenético con aplicaciones de la biología molecular. Mediante esta técnica una sonda marcada correspondiente a un fragmento de DNA específico se híbrida con los cromosomas correspondientes en metafase, profase o interfase. Posteriormente se puede identificar la sonda mediante un microscopio para inmunofluorescencia. Esta técnica de citogenética molecular nos permite reconocer microdeleciones cromosómicas que no se pueden detectar con las técnicas de bandeo de alta resolución1.
Si nuestro caso no pertenece al grupo de enfermedades debidas a una alteración cromosómica, se sospecha que presenta un patrón de herencia mendeliano y no se trata de una enfermedad en la que está estandarizado el estudio molecular, es necesario recurrir a las diferentes bases de datos genéticas para conocer si se ha localizado el locus, si se ha identificado el gen causante, el número de mutaciones y la prevalencia de cada una de ellas. Las alteraciones cognitivas afectan al 5-7 por ciento de la población, habiéndose descrito múltiples enfermedades genéticas capaces de alterar mecanismos implicados en memoria y aprendizaje, condicionando defectos cognitivos similares. En las bases de datos de enfermedades debidas a defectos en un único gen, son múltiples las alteraciones de aprendizaje aisladas o asociadas a diferentes fenotipos clínicos –en el año 2008 en OMIM (Online Medelian Inherited in Men) se registran más de 1.000 entradas diferentes que condicionan retraso mental–. Además de las alteraciones cognitivas relacionadas con alteraciones cromosómicas, metabólicas o asociadas a algún tipo de neuropatía, existen otras presumiblemente ligadas a problemas del desarrollo y en muchas ocasiones sin causa conocida12.
Únicamente podremos iniciar un estudio molecular dirigido cuando se sospeche una mutación determinada, bien por su prevalencia o bien por los datos de la semiología del paciente cuando existe una adecuada correlación genotipo-fenotipo. Cuando el número de mutaciones descritas es muy abundante, en ocasiones puede recurrirse al estudio indirecto de marcadores. Si el locus implicado ha sido localizado, pero el gen no ha sido caracterizado únicamente podemos realizar esta estrategia de diagnóstico indirecto basada en la utilización de marcadores polimórficos próximos al locus. Esta información tiene una alta calidad informativa a pesar de que se trata de una estrategia probabilística. En todo caso, una vez realizado el estudio genético, trataremos de identificar el patrón de segregación familiar, que aportará criterios de riesgo de ocurrencia o recurrencia en esa familia. Si el patrón de herencia identificado es autosómico recesivo, la existencia de un hijo afectado nos indica que los padres son heterocigotos y por lo tanto la probabilidad de que cada nuevo descendiente esté afectado será de un 25%. Si el patrón de herencia es autosómico dominante, la probabilidad de que un feto esté afectado será de un 50%. Finalmente si presenta una herencia ligada al cromosoma X y el padre esta afectado, todas sus hijas serán heterocigotas y todos sus hijos serán normales; si la madre es portadora, sus hijas tendrán una probabilidad del 50% de ser normales o portadoras y sus hijos una probabilidad del 50% de ser normales o afectados.
Estas estrategias podrán ser aplicadas en la detección y caracterización de una enfermedad hereditaria en distintas etapas del desarrollo o bien a distintos colectivos de enfermos, definiéndose los distintos tipos de diagnóstico genético. Así por ejemplo, en función de la etapa del desarrollo del individuo analizado tendremos un diagnóstico preimplantacional, cuando se analiza el genotipo en las primeras etapas embrionarias con el objeto de detectar una determinada alteración genética; realizaremos un diagnóstico prenatal cuando el genotipo es analizado durante las primeras semanas de desarrollo fetal; un diagnóstico postnatal cuando el genotipo se determina después del nacimiento, normalmente durante los primeros días después del parto (neonatal) o en períodos posteriores de la infancia o de adulto.
Patología prenatal
Cada vez existe mayor interés por los factores que pueden afectar al embrión y al feto, ya que los problemas del crecimiento y desarrollo del nuevo ser, desde el momento de la concepción hasta el nacimiento, tienen una gran trascendencia en la vida futura del niño y del adulto. Aunque la medicina prenatal es un campo de investigación que interesa a distintas áreas de la medicina (genetistas, embriólogos, patólogos, obstetras, etc.), son los pediatras los más interesados en el tema desde el punto de vista práctico, pues deberán cuidar y atender las anomalías que durante este periodo se presenten o al menos tratar de conocer los mecanismos que las producen para evitar el anormal desarrollo del embrión y del feto.
El término patología prenatal es extraordinariamente amplio, ya que en él queda englobada toda la patología que va desde el momento de la concepción hasta el nacimiento. Durante este periodo pueden actuar en sentido patológico tanto factores genéticos como ambientales. Esta patología tiene una enorme importancia en pediatría ya que, como hemos comentado, son una causa importante de mortalidad infantil y a menudo producen discapacidad en el niño que los padece.
El conocimiento que se va teniendo sobre el origen y los mecanismos de producción de las anomalías prenatales ha inducido a procurar un diagnóstico prenatal en muchas de ellas, con afanes preventivos unas veces a través del aborto, o con ideas de aplicación precoz de una terapéutica en otros. Aunque se ha progresado bastante en este terreno, debe tenerse presente que aún no es posible, ni mucho menos, detectar la mayoría de las anomalías conocidas.
Citogenética y diagnóstico prenatal
Los años cincuenta fueron testigo del desarrollo de la citogenética, el estudio de los cromosomas al microscopio. Su uso diagnóstico pronto reveló que las anormalidades en el número de cromosomas –generalmente uno de más o uno de menos– causaban invariablemente una disfunción profunda. Los problemas se derivan de un desequilibrio en el número de genes, una desviación de la norma que es tener dos ejemplares de cada cromosoma. Estas enfermedades no se repiten en una misma familia como ocurre en las enfermedades por defectos en un único gen como la fenilcetonuria, pero también tienen mucho de genéticas: se producen de manera espontánea por accidentes en la división celular que da origen a los espermatozoides y a los óvulos. La más conocida de estas enfermedades es el síndrome de Down, enfermedad que en el lenguaje genético se conoce como trisomía del cromosoma 21, cuya incidencia aumenta con la edad de la madre. A los veinte años la probabilidad de engendrar un bebé con el síndrome de Down es 1 entre 1.700; pero a los treinta y cinco salta a 1 entre 400 y a los cuarenta y cinco se dispara a 1 entre 30. Por esta razón, muchas embarazadas tardías optan por el diagnóstico prenatal del feto para determinar si tiene triplicado el cromosoma 21. La prueba se hizo por primera vez en el año 1968 y hoy es un procedimiento diagnóstico de rutina para todas las embarazadas mayores de 35 años. Dado que el feto tiene que ser lo suficientemente grande como para que la extracción de la muestra de tejido sea segura, esta prueba diagnóstica no debe realizarse en los primeros momentos del embarazo. Normalmente se lleva a cabo entre la semana quince y la dieciocho mediante amniocentesis, un procedimiento de extracción de líquido amniótico que contiene de forma natural células del feto. Existe una prueba alternativa, pero menos fiable, que se puede hacer desde la décima semana de embarazo y que consiste en tomar células del corion, la parte de la placenta que se sitúa junto a la pared del útero. Como los dos procedimientos tienen un cierto riesgoc, se suele aconsejar a las mujeres jóvenes que no se sometan a estos procedimientos pues la probabilidad de que sus fetos tengan un defecto genético es mucho menor que la de que resulten dañados por el propio procedimiento. Hace años, antes de procesarlas para hacer el análisis cromosómico las células fetales extraídas tenían que ser cultivadas en el laboratorio. Hoy día, se puede hacer un diagnóstico más rápido con la hibridación fluorescente in situ. Este método consiste en ligar una pequeña molécula fluorescente a un fragmento de ADN específico del cromosoma 21 que posteriormente se introduce en la muestra, donde se unirá al cromosoma 21 del feto. Si aparecen dos manchas fluorescentes en el núcleo de la célula el feto es normal; si hay tres el feto tiene el síndrome de Down13.
En nuestro entorno, se detecta el 30 por ciento de los embarazos con síndrome de Down mediante la realización rutinaria de la prueba a las mujeres embarazadas de más edad –un cinco por ciento del total–. Muchos estudios han demostrado que este método es claramente eficaz en términos de detecciones por cada euro invertido, pero el 70 por ciento de los casos de Down no se detectan. El síndrome de Down es menos frecuente en los bebés de las madres jóvenes, pero esas mujeres son las que tienen el mayor número de embarazos, y en ellas las pruebas diagnósticas de rutina no compensan estadísticamente el riesgo concomitante que provocan, por lo que se ha intentado encontrar indicadores alternativos que no sean invasivos y a este respecto se ha observado que existen sustancias detectables en la sangre de la madre que pueden proporcionar información útil. Unos niveles bajos de alfa-fetoproteína y elevados de gonadotropina coriónica presentan una correlación significativa con el síndrome de Down, aunque de ningún modo son indicadores definitivos de la trisomía. En la actualidad es práctica habitual ofrecer el test sanguíneo a las embarazadas jóvenes, y, si éste sugiere la posibilidad de un Down, se les aconseja que se sometan a una amniocentesis o a la toma de una muestra de células del corion para obtener un diagnóstico definitivo.
En nuestro entorno las mujeres que conocen que tienen un feto con Down deciden interrumpir el embarazo hasta en el 90 por ciento de los casos. Lo habitual es que únicamente las mujeres que están dispuestas a abortar se sometan al diagnóstico prenatal (no tiene sentido exponer al feto a los riesgos asociados con el procedimiento, si la madre tiene intención de llevar su embarazo a término sea cual sea el resultado), por lo tanto es lógico que esta cifra sea elevada. El resultado es que en los países en los que se lleva a cabo un cribado prenatal de rutina, la incidencia de nacidos con síndrome de Down desciende hasta en un 40 por ciento. La determinación del impacto de esta prueba sobre la incidencia de esta alteración cromosómica es difícil de obtener debido a la tendencia a posponer la maternidad, lo cual incrementa el número de mujeres con riesgo de tener un embarazo con síndrome de Down. Por eso en los países más desarrollados, la eficacia de los programas de cribado se mide en relación con el número esperado de bebés con Down, teniendo en cuenta la edad de las mujeres que tienen hijos en un año determinado 14.
Las trisomías pueden ocurrir también en otros cromosomas, pero provocan unas alteraciones tan graves que esos embarazos terminan en abortos espontáneos en todos los casos excepto en las trisomías de los cromosomas 13 y 18: los niños con trisomía del 13 rara vez viven más de unas pocas semanas y los que presentan la trisomía del 18 generalmente mueren antes de cumplir un año. Las anormalidades cromosómicas, incluidas las trisomías, son seguramente muy comunes, y mientras que algunas son letales –la estimación actual es de que hasta el 20 por ciento de las concepciones terminan en abortos espontáneos y que en más o menos la mitad de ellas existe algún tipo de aberración cromosómica–, otras tienen poco o ningún efecto. Las alteraciones pueden ser menos drásticas que la pérdida o ganancia de un cromosoma completo y pueden implicar reordenamientos de segmentos dentro de un cromosoma o la transferencia de una parte de un cromosoma a otro. Si ha habido una pérdida o ganancia neta de material genético, entonces, como en el caso de un cromosoma completo extra, el desequilibrio resultante será probablemente dañino. Por desgracia, los análisis citológicos normales de los cromosomas fetales pueden detectar solamente grandes desequilibrios, y sin embargo, incluso los más pequeños pueden tener efectos devastadores.
Diagnóstico pre-implantación
El diagnóstico preimplantatorio permite una intervención antes del reconocimiento clínico del embarazo y evitar por lo tanto la interrupción clínica del mismo. Esto lo convierte en una opción atractiva para las parejas que no pueden resolver su conflicto ético con el aborto o que tienen un riesgo excepcionalmente alto para un trastorno genético. Combinando dos tecnologías vanguardistas –la fertilización in vitro (FIV) y el diagnóstico del ADN basado en la PCR– es posible analizar la situación genética de un embrión antes de ser implantado en el útero de una mujer y empezar su desarrollo. Después de la fertilización in vitro, los productos de la concepción se cultivan en el laboratorio hasta que cada óvulo fertilizado se ha dividido tres o cuatro veces produciendo un conjunto de ocho a dieciséis células. Se toman una o dos células de cada uno para extraer ADN y se utiliza la PCR para amplificar las secuencias relevantes, todo ello con objeto de determinar en cada caso si hay o no alguna mutación. La capacidad que tiene la PCR de amplificar cantidades mínimas del ADN que se intenta localizar es la que hace que sea posible este método de diagnóstico precoz. Los padres tienen entonces la libertad de implantar sólo los embriones que son negativos para la enfermedad genética.
Las primeras pruebas diagnósticas preimplantacionales se realizaron en 1989 para examinar el sexo del feto, una información muy importante cuando el riesgo es padecer una enfermedad ligada al sexo. Una madre portadora puede seleccionar exclusivamente embriones femeninos partiendo de la base de que no padecerán la enfermedad, aunque puedan ser portadoras. El diagnóstico preimplantacional puede ampliarse más allá de la simple determinación del sexo, con el objetivo de llegar a detectar mutaciones específicas.
En nuestra cultura, la biología reproductiva humana parece ser una fuente inacabable de polémica, y es seguro que cualquier procedimiento que implique manipulación de embriones humanos con cualquier propósito se convertirá en foco de controversia. El diagnóstico preimplantacional no ha sido una excepción. Dejando a un lado las consideraciones éticas, el procedimiento sigue teniendo hoy por hoy dos inconvenientes principales: exige un alto grado de compromiso de la pareja a la que se le realiza y, como todas las variantes de la fertilización in vitro es muy caro. El diagnóstico preimplantacional tiene el potencial de convertirse en un arma importante en la batalla contra la enfermedad genética y la discapacidad en familias con historia previa de una enfermedad hereditaria determinada.
¿Es posible un tratamiento?
Muchas de las enfermedades genéticas aparecen con una desconsoladora frustración: sabemos lo suficiente como para diagnosticarla, quizá para eludirla, pero no para tratarla. Afortunadamente, existen algunos casos en los que el conocimiento genético nos ha hecho avanzar en el camino, aportando terapias que curan. Pero, por desgracia, pocos remedios resultan sencillos y efectivos como el de la fenilcetonuria, enfermedad en la que los afectados pueden recobrar una vida normal con unas pocas restricciones en la dieta.
Demasiado a menudo los trastornos genéticos diezman las células de un tejido concreto: los músculos en la enfermedad de Duchenne, las células nerviosas en el Huntington y el Alzheimer, no existiendo una solución fácil para este tipo de deterioro insidioso. Existe una posibilidad real de que en un futuro seamos capaces de tratar ese tipo de enfermedades usando células madre. Muchas de las células del cuerpo sólo son capaces de reproducirse a sí mismas –una célula hepática, por ejemplo, sólo produce células hepáticas–; pero las células madre pueden generar una variedad de tipos especializados de células. En el caso más sencillo, un óvulo recién fecundado –la célula madre con el máximo potencial–, originará en último extremo cada uno de los doscientos dieciséis tipos de células humanas reconocidas. La manera más natural de encontrar células madre es en los embriones; también se pueden encontrar en los adultos, pero esas células suelen carecer de la habilidad embrionaria para diferenciarse en cualquier tipo de célula. Comenzamos ahora a entender cómo inducir a las células madre para que produzcan un tipo de célula específico y algún día será posible compensar con células nuevas las pérdidas de células en el cerebro de las personas con Huntington o Alzheimer, pero queda mucho camino por recorrer. Pasarán unos diez años antes de que nos encontremos en situación de aplicar adecuadamente el posible valor terapéutico de las células madre. Actualmente la restrictiva legislación a nivel mundial está obstaculizando los esfuerzos para desarrollar esta tecnología potencialmente valiosa, debiéndose reconocer, por otra parte, la necesidad de poner ciertos límites a la manipulación genética.
En el momento actual no es posible reemplazar en una enfermedad genética el total de las células alteradas mediante la terapia con células madre, pero sí que puede reemplazarse una proteína ausente. El tratamiento de muchas enfermedades no combate la raíz genética de la enfermedad, pero minimiza el efecto de la mutación aportando al paciente la proteína vital que el gen alterado no puede aportar. Evidentemente es posible y eficaz enderezar la anomalía genética a través de recurso bioquímico, pero incluso considerando la notable eficacia de los métodos recombinantes, los tratamientos son caros y suponen una gran dependencia del sistema sanitario. Por eso hace tiempo que los genetistas sueñan con una forma práctica de resolver el problema más que de compensar sus efectos. El tratamiento ideal de una enfermedad genética sería hacer algún tipo de modificación genética que permitiera corregir los genes que causan el problema y que esa corrección durara de por vida. Hay dos procedimientos, al menos en principio: terapia génica en células somáticas, mediante la cual se modificarían los genes de las células del cuerpo del paciente; y terapia génica en línea germinal, mediante la que se modificarían los genes de los espermatozoides o de los óvulos del paciente, evitando la transmisión de la mutación dañina a la generación siguiente.
Este tipo de soluciones teóricas a los estragos causados por los defectos genéticos puede parecer obvio, pero la idea de la terapia génica no ha sido recibida con demasiado entusiasmo ni por los profesionales ni por la población. La reacción no es del todo sorprendente: una cultura preocupada por la modificación de una planta de maíz es de esperar que esté en contra de personas transgénicas –o de seres humanos modificados genéticamente, si se prefiere– a pesar de los potenciales beneficios. En la terapia génica somática, el efecto de ese daño puede ser limitado; pero en la terapia génica germinal existe la posibilidad de producir de forma accidental individuos deficientes. Incluso sus defensores no sugerirían que este tipo de procedimiento se lleve a cabo hasta que nuestras técnicas no sean lo suficientemente buenas como para confiar en que no se causará ningún daño inadvertidamente. No obstante, la controversia es por ahora puramente académica, pues la terapia génica germinal está muy por encima de nuestras posibilidades técnicas, y hasta que no esté a nuestro alcance, deberíamos concentrarnos en conseguir que la terapia génica somática se convierta en una herramienta poderosa por sí misma 15.
El diagnóstico prenatal como camino para nuevas opciones terapéuticas
Los procedimientos para detectar trastornos genéticos en reproducción humana son cada vez más variados y más precoces y el feto es cada vez más accesible, no sólo para el diagnóstico precoz sino también para el tratamiento. De hecho puede accederse al embrión incluso antes de su implantación (diagnóstico pre-implantatorio), e incluso es posible obtener información antes de su concepción (biopsia del primer cuerpo polar) y no sólo eso, sino que se puede obtener material genético fetal sin procedimientos invasivos para el feto (células fetales en sangre materna, mucus transcervical). Merece la pena, por tanto, repasar aunque sea brevemente estos avances y su posible repercusión en relación a las enfermedades que causan grave discapacidad.
La tecnología para el diagnóstico avanza más velozmente que las posibilidades terapéuticas, pero esto no significa que la terapia in útero no sea objetivo para la medicina. La terapia génica fetal ciertamente está en fase experimental en modelos animales pero desde el punto de vista teórico tiene características interesantes: posibilidad de dirigirla a células madre en expansión y a órganos o sistemas celulares que serán inaccesibles más adelante, conferir una expresión estable del gen y evitar la sensibilización inmunitaria frente al producto transgénico. La investigación en modelos animales dará lugar además a nuevas aproximaciones al conocimiento de la biología del desarrollo.
Aún así, el diagnóstico genético pre-implantatorio elimina embriones, puesto que los diagnosticados afectados no son implantados. La única manera de evitarlo sería establecer el diagnóstico en gametos (oocitos y espermatozoides) antes de la fertilización, es decir, hacer un diagnóstico preconcepcional, con lo cual se eliminan los gametos con el defecto genético.
Expectativas en la prevención y tratamiento
Existen pocas medidas de prevención de las enfermedades genéticas, fuera del consejo genético para evitar embarazos de alto riesgo o la detección antenatal de los individuos afectados. Los controles de salud durante el embarazo y el acceso a las técnicas de diagnóstico prenatal son las únicas medidas eficaces. El diagnóstico precoz del caso índice, además de conducir al inicio de programas de atención optimizados y el acceso a posibles medidas terapéuticas, conlleva siempre la posibilidad de establecer opciones preventivas para la familia, en primer lugar mediante el consejo genético, que suele hacerse extensivo, previo consentimiento de los padres, a otros miembros de la familia, de manera que cada caso suele desencadenar lo que se denomina un cribado en cascada, empezando por la búsqueda de los pacientes todavía asintomáticos (si van a beneficiarse con ello de una mejora en su calidad de vida) y la de heterocigotos, que es muy relevante, especialmente en las enfermedades ligadas al cromosoma X. El consejo genético contemplará asimismo la información sobre las opciones reproductivas, incluyendo las posibilidades de la técnica de reproducción asistida a las que nos hemos referido.
No cabe duda de que en el futuro el tratamiento genético será el tratamiento de elección en un número cada vez mayor de casos de problemas genéticos, pero por el momento las dificultades técnicas derivadas de la necesidad de encontrar mejores y más seguros vectores han retrasado el ritmo de incorporación de estas opciones terapéuticas más de lo previsto. La terapia génica somática mediante administración del gen deficiente ya sea con técnicas in vivo, administrando el ácido nucleico terapéutico mediante un vector de empaquetamiento, o ex vivo, administrando mediante células del propio paciente previamente extraídas y manipuladas, está particularmente indicado en los casos de deficiencia de un enzima catalítico, donde una pequeña recuperación de la actividad enzimática puede restaurar la normalidad terapéutica. La manipulación de la expresión genética y la inhibición de la síntesis del DNA anormal representan estrategias alternativas que seguramente mejorarán el pronóstico de muchas enfermedades, pero en la actualidad no pueden ser tenidas en cuenta como tratamiento de los pacientes con enfermedades genéticas complejas.
Ambos, la prevención y el tratamiento eficaz, forman parte de los principales objetivos médicos de cualquier enfermedad, y por tanto afectan a las enfermedades de base genética, teniendo en cuenta las limitaciones que la terapia génica tiene hasta la fecha. Señalamos de nuevo la necesidad de profundizar en los mecanismos últimos que condicionan la discapacidad intelectual de personas con enfermedades genéticas complejas, en las que están afectados de forma simultánea y permanente diferentes genes o regiones genéticas reguladoras del desarrollo o de las respuestas del individuo ante situaciones ambientales determinadas, y sobre las que podían implementarse medidas más realistas que las basadas en el intercambio de genes. Resulta asimismo necesario que los médicos responsables de los pacientes con enfermedades de base genética posean los conocimientos y habilidades suficientes para poder establecer las medidas terapéuticas generales, que permitan la supervivencia de los pacientes sospechosos de padecer una enfermedad mejorable desde el punto de vista médico, como son los errores congénitos del metabolismo; organizar el tratamiento sindrómico más adecuado en función del perfil clínico y bioquímico del niño, conocer las posibilidades terapéuticas de los enfermos de cuyo cuidado general es responsable y ser capaz de prevenir, y detectar en su caso, las situaciones de riesgo para cada enfermedad. Todo ello, con el objetivo de lograr que el enfermo puede llevar una vida lo más normal posible, debidamente integrado en su medio familiar, escolar o laboral.
Disección de los síndromes genéticos
Muchas de las enfermedades a las que nos hemos referido son simples desde el punto de vista genético: están causadas por una mutación en un único gen y el entorno no influye en si padecerá o no la enfermedad. La situación es mucho más complicada en el caso de la mayoría de las enfermedades ligadas al desarrollo condicionantes de discapacidad intelectual, las cuales pueden ser desencadenadas por una combinación de factores hereditarios y ambientales, fundamentalmente factores prenatales. Pero incluso en las enfermedades complejas, hay unos cuantos genes específicos como los factores de regulación transcripcional, que, con independencia del ambiente, tienen un efecto decisivo y sobre las que es necesario investigar.
Analizaremos a continuación alguno de los aspectos que consideramos relevante desde el punto de vista que hemos denominado de disección de la lesión genética. La pérdida de una región cromosómica condiciona que un individuo tenga una única copia de una porción del genoma, con la consiguiente hemicigosidad o haploinsuficiencia para uno o varios locus genéticos. Las consecuencias clínicas dependen del tamaño de la zona perdida y del número y función de los genes contenidos en ella. Las pérdidas de información genética pueden ser nuevas (de novo) o pueden ser hereditarias como consecuencia de una mala segregación de una traslocación balanceada en sus progenitores. Además de las pérdidas de material genético (deleciones) que son fácilmente detectables mediante estudios citogenéticos, las nuevas técnicas de cariotipado de alta resolución y técnicas de citogenética permiten identificar mínimas pérdidas de material en un porcentaje significativo de pacientes con síndromes únicamente reconocibles desde el punto de vista clínico hasta hace pocos años, al presentar pérdidas submicroscópicas de material genético. Asimismo, en muchas enfermedades metabólicas el desarrollo de las técnicas de análisis genético ha permitido mejorar el diagnóstico, especialmente el diagnóstico prenatal y de portadores, al poder determinar con rapidez, precisión y fiabilidad si un individuo porta un alelo mutante. El análisis genético también puede aplicarse en la predicción de la evolución clínica y en la orientación del tratamiento, ya que, en algunos casos, se ha podido relacionar la mutación con el grado de severidad de la enfermedad.
La pérdida de la región cromosómica o defecto cromosómico asociado con cada síndrome, aunque sea submicroscópica, contiene múltiples genes y es esperable que las manifestaciones clínicas que clásicamente definen el síndrome sean el resultado de la pérdida de los diferentes genes situados en la misma región cromosómica. Clínicamente, estas enfermedades se caracterizan por anomalías de múltiples sistemas y cada uno de ellos se ha podido asociar a defectos citogenéticos o moleculares en un elevado porcentaje de pacientes. No es probable que todos y cada uno de los genes afectados por el defecto genético juegue un papel importante en las manifestaciones mayores de cada fenotipo clínico o síndrome, por tanto, sería más apropiado definir cada uno de estos síndromes como una condición en la cual numerosos genes contiguos pueden tener un efecto dependiente de dosis, entre los que un mínimo número contribuye a las características propias del fenotipo. Alguna de estas enfermedades pueden finalmente ser definidas por un defecto en único gen cuando se analicen todos los genes en la región afectada, abriendo nuevas perspectivas terapéuticas11.
Hay estudio del perfil metabólico
Para iniciar la investigación diagnóstica, el nivel genético es útil como punto de partida para enfermedades con características clínicas muy bien definidas y lo bastante específicas como para obtener el diagnóstico. Debemos, sin embargo, tratar de avanzar y aproximarnos al diagnóstico metabólico o de la función de los déficits genéticos. El diagnóstico molecular de las enfermedades hereditarias puede ser abordado desde un nivel genético, analizando el producto génico o proteína alterados, pero también desde las vías metabólicas afectadas a través de la determinación de metabolitos relevantes. Una vez considerado que la terapia génica a día de hoy no es una opción terapéutica al alcance de los pacientes que sufren una enfermedad de base genética, debemos establecer estrategias de investigación en busca de nuevas opciones terapéuticas. Cuando carecemos de recursos de tratamiento específicos o el cuadro clínico que motiva una investigación molecular es poco específico, conviene alejarse del gen, siendo a menudo la mejor aproximación a nivel metabólica con el fin de obtener información acerca del funcionamiento de muchos genes a la vez. La identificación de un perfil metabólico en un paciente concreto y en una situación determinada permitirá además reconocer los cambios biológicos que aparecen tras los tratamientos establecidos, tanto farmacológicos como psicoeducativos. Es de suponer que un cambio conductual como resultado de un programa de intervención se acompañe de cambios biológicos que pudieran objetivarse de algún modo mediante la monitorización de perfiles metabólicos determinados. La importancia de la detección precoz de muchos de estos déficit radica en que el diagnóstico y tratamiento adecuado en periodos críticos del desarrollo modifica la historia natural de las mismas evitando afectación severa de los individuos que las padecen 16.
Genética del comportamiento
Algunos de los descubrimientos más importantes y recientes sobre comportamiento están relacionados con la genética. Así, por ejemplo, el autismo es un trastorno raro pero grave en la infancia. Los niños autistas se retraen socialmente, no tienen contacto físico o visual, con un marcado déficit de comunicación y un comportamiento estereotipado. Hasta la década de los ochenta, se pensaba que el autismo tenía una causa ambiental o infecciosa, un rechazo de los padres o una lesión cerebral. Estudios genéticos realizados comparando el riesgo de gemelos idénticos (monocigóticos), los cuales son genéticamente iguales (como clones), y gemelos dicigóticos mellizos, que sólo tienen la mitad de su dotación genética en común, han señalado una notable influencia genética. Si un miembro de la pareja de gemelos monocigóticos (MZ) es autista, el riesgo de que el otro gemelo sea autista es del 60%. En el caso de los mellizos o gemelos dicigóticos (DZ), el riesgo es del 10%. Se ha emprendido una serie de estudios de genética molecular con el fin de identificar genes individuales que contribuyan a la susceptibilidad genética de padecer autismo.
La investigación genética del comportamiento no sólo demuestra la importancia de la genética en este campo, sino que va mas allá y nos permite plantear cuestiones acerca de cómo los genes influyen en el comportamiento. En este sentido, ¿varía la influencia genética durante el desarrollo?; consideremos la habilidad cognitiva por ejemplo. Se podría pensar que las diferencias ambientales irían aumentando a lo largo de nuestra vida mientras que las diferencias genéticas podrían ser menos importantes. No obstante, la investigación genética demuestra justo lo contrario; la influencia genética en la habilidad cognitiva crece a lo largo de la vida de un individuo, alcanzando niveles en las últimas etapas de la vida tan importantes como la influencia que tienen los factores genéticos en la estatura. Este descubrimiento constituye un ejemplo del análisis genético del desarrollo.
El éxito escolar y los resultados de las pruebas que se realizan en la escuela están influidos casi en igual manera por la genética como lo están los resultados de los tests de capacidades cognitivas tales como los tests de inteligencia (CI). Aún más interesante, el considerable solapamiento que se produce entre dicho éxito escolar y la capacidad de realizar bien los exámenes es casi todo de origen genético y éste sería un ejemplo de lo que se denomina análisis genético multivariante.
La investigación genética también está cambiando el modo de pensar acerca del ambiente. Por ejemplo, se suele pensar que el crecer en el seno de una misma familia hace que los hermanos y hermanas sean psicológicamente parecidos. Sin embargo, en la mayoría de los aspectos y trastornos del comportamiento, es la genética la que explica la similitud entre hermanos. Aun cuando el entorno es importante, las influencias ambientales hacen que los hermanos que crecen dentro de la misma familia sean diferentes, y no iguales. Este tipo de investigaciones ha desencadenado una explosión de estudios del ambiente que buscan las razones ambientales por las cuales los hermanos de una misma familia son tan diferentes.
Avances moleculares y farmacogenética
El desarrollo espectacular de la Biología Molecular durante los últimos años ha conducido, no sólo al conocimiento a escala molecular del funcionamiento normal de los procesos que mantienen la vida, sino también al entendimiento del origen de ciertas enfermedades cuya etiología no sería posible conocer de otro modo. Es cierto que la Bioquímica, mediante el estudio del metabolismo y de las enzimas que lo regulan, ha abierto el camino, acuñando, incluso, términos tales como enzimopatías o errores del metabolismo, sobre los que hoy se sostiene la Patología Molecular. Sin embargo, han sido las técnicas de biología molecular las que han dado el impulso definitivo para el desarrollo de la Patología Molecular, que ha permitido acercarse directamente al genoma para buscar con detalle el origen molecular de las enfermedades. Así, las técnicas de biología molecular permitieron, primero, localizar las enfermedades en sus respectivos cromosomas y, más tarde, la descripción formal de las mutaciones implicadas.
La identificación de la etiología molecular de las enfermedades está lejos de tener un interés puramente académico. El conocimiento de las mutaciones individuales ha dado paso a nuevas a disciplinas como la farmacogenética que intenta diseñar fármacos específicos a la idiosincrasia genética de cada enfermo. Este diseño a medida de los fármacos requiere un conocimiento profundo de los genes implicados en la enfermedad, así como de sus posibles polimorfismos o variantes genéticas que condicionan un fenotipo individual para cada enfermedad. Por consiguiente, es necesario desarrollar al máximo la Patología Molecular con objeto de llevar a cabo, no sólo el diagnóstico preciso de la enfermedad, sino también el diseño de la estrategia de su posible curación basada en el conocimiento íntimo de los genes implicados, ya sean genes afectados o sanos. En el futuro, pues, la Patología Molecular no se limitará al mero estudio del gen o los genes responsables de la enfermedad, sino que necesitará la localización de los polimorfismos de otros genes de cuya interacción con el gen o genes afectados depende el grado de morbilidad de la enfermedad17.
Perspectivas futuras en el análisis molecular
Es virtualmente imposible el conocimiento de todos los síndromes y enfermedades genéticas, pero como hemos señalado, es necesario aproximarse a los posibles defectos genéticos que condicionan una enfermedad para diseñar estrategias preventivas y terapéuticas. En este sentido, el manejo de diversos catálogos y bases de datos facilita la búsqueda de genes responsables de enfermedades y pueden ser la clave para definir diversas entidades patológicas.
En la actualidad, aunque disponemos de importantes fuentes bibliográficas para la búsqueda de genes responsables de enfermedades y se conoce la metodología adecuada para identificar en muchos casos dichos genes, en la práctica resulta complejo y muy costoso obtener el diagnóstico molecular de la mayoría de las enfermedades genéticas. Habitualmente deberemos recurrir a laboratorios de referencia para este fin y puede ser muy difícil obtener un diagnóstico molecular en aquellas enfermedades en las que el procedimiento no esté estandarizado. No obstante, en el momento actual asistimos al desarrollo tecnológico de las denominadas micromatrices de ADN, que significarán en un próximo futuro una excelente herramienta para obtener diagnósticos más rápidos y exactos de muchas enfermedades, así como para facilitar la individualización de los tratamientos clínicos. Las micromatrices o microchips de investigación genotípica, capaces de identificar simultáneamente miles de rasgos moleculares, pueden tener aplicaciones muy diversas que van desde la comparación de genes de organismos diferentes (por ejemplo, en la búsqueda de claves sobre la historia de la evolución de los organismos, o la comparación de genes en modelos animales de disfunción cognitiva) hasta la comparación de los genes de candidatos a participar en los procesos cognitivos de individuos normales y deficientes, en busca de sutiles diferencias de composición o número de genes, e incluso permitir identificar defectos cognitivos a nivel molecular.
Como comentario final, podemos añadir que, aunque para la mayoría de los ciudadanos en general, y los médicos en particular, ni la genética molecular ni la biología molecular o celular han intervenido ni intervienen en la práctica clínica del día a día, a medida que mejore nuestra comprensión de las células y del organismo entero gracias a los avances en el conocimiento y el desarrollo tecnológico, los médicos podrán diagnosticar con mayor precisión, indicar terapias más depuradas (incluidas probablemente las genéticas) y ajustar sus tratamientos a las características constitucionales y al estado fisiológico real de cada paciente. Esta explosión de conocimiento e información ha sido desencadenada por una serie de avances tecnológicos y avivada por el progreso del Proyecto Genoma Humano. La conjunción del arte de la medicina y de la precisión de la ciencia ha modelado una nueva disciplina –medicina molecular– que vislumbra el control de muchos aspectos de las enfermedades de base genética, siendo la Biología Molecular el paradigma de la práctica y de la enseñanza médicas. Sería una magnífica herencia para la medicina del futuro que hacia el año 2020, las instituciones sanitarias dispusieran de múltiples modelos del estado molecular de sus pacientes y simulaciones virtuales que podrían actualizarse constantemente, que aporten nuevos niveles de comprensión en el campo de la biología molecular y celular, y de este modo poder prevenir aquellas enfermedades discapacitantes de base genética mediante la anticipación en el caso de padres portadores de mutaciones determinadas, identificación temprana de los individuos con riesgo o mediante el diseño de estrategias terapéuticas específicas aplicadas de manera precoz. La medicina molecular introducirá en el contrato clínico una forma sin precedentes de pronóstico; juicio que jugará un papel protagonista en numerosas situaciones como es la identificación posible de alteraciones genéticas en las primeras etapas del desarrollo embrionario y que se vislumbra, en pocos años, como un estudio de rutina.
Nos enfrentaremos a la difícil definición del estado creado por la presencia de tales genes defectuosos que identifica un estado que podríamos denominar de susceptibilidad, predisposición, propensión, proclividad, riesgo potencial o probabilidad. Independientemente del término elegido para designar una situación concreta en cada individuo, la información genética implica la toma de decisiones que pueden afectar la viabilidad de un embrión con un gen deletéreo para su futuro como adulto, o cómo manejar la información que afecta a un paciente o a sus familiares, respecto al riesgo de sufrir una enfermedad discapacitante –más o menos grave, en un momento u otro de su vida–, con alguna probabilidad en algunos casos y en otros casos con certeza.
Consejo genético
La meta final de la detección de un síndrome hereditario y de la determinación de un test genético es la realización de un adecuado consejo genético, en el caso que esto sea posible. Pero, ¿qué es realmente el consejo genético? Una definición posible sería la de proceso por el cual se informa a los pacientes, y familiares del mismo, de la posibilidad de padecer una enfermedad hereditaria, de la posibilidad de transmitirlo a las siguientes generaciones, de las medidas preventivas y terapéuticas que se pueden realizar así como de la posibilidad de realizar un test genético. Efectivamente, el consejo genético no engloba obligatoriamente la realización de un test genético (de hecho, en muy pocos casos podremos realizarlo y, en menos casos todavía, la información será lo suficientemente clara como para que podamos tomar alguna decisión en función del resultado).
La función de la consulta de consejo genético18 es la valoración del riesgo que existe en una determinada familia con criterios de padecer una enfermedad hereditaria de que cada individuo de la misma pueda padecer un determinado tipo de enfermedad. Los avances moleculares de los últimos años nos van a permitir realizar estudios de mutaciones en alguno de estos síndromes pero en otros casos no; en esta segunda situación podremos hacer una valoración aproximada del riesgo en función de la historia familiar y los factores exógenos a los que cada individuo de esta familia se ve sometido.
Cuando ya se ha valorado este riesgo y se comprueba que está por encima del asumido por la población general se le deben comentar a cada sujeto los beneficios y perjuicios de conocer esta información, tanto desde el punto de vista médico como psicológico y social, así como las posibilidades de manejo de esta situación haciendo hincapié en las limitaciones de las mismas. En el caso de que exista un test genético adecuado a ese caso en concreto, deberemos explicar a la persona que consulta las posibilidades que existen de que el resultado sea positivo, negativo o no informativo, así como las implicaciones que tendría cada una de ellas.
Si tras esta información el sujeto sigue interesado en recibir consejo genético procederemos a realizar un test que suele consistir en una extracción de sangre de donde extraeremos el ADN linfocitario para el estudio de mutaciones en el gen o genes probablemente afectos. Previo a esta extracción el paciente ha debido firmar un consentimiento informado donde se recogen varios puntos que van desde la información específica sobre el test que se va a realizar; las implicaciones del tipo de resultado que se obtenga; las posibilidades de que el test sea negativo, positivo o no informativo; las opciones de valorar el riesgo sin necesidad de acudir a este tipo de pruebas; la posibilidad de transmisión a la descendencia; la fiabilidad técnica de la prueba; los riesgos de distrés emocional; el riesgo de implicaciones laborales o de seguros así como de las limitaciones que presenta el manejo médico de las mismas. Tras la firma de dicho consentimiento se realiza la extracción sanguínea y, tras un tiempo de espera hasta obtener el resultado (en algunos casos pueden ser varios meses), deberemos proceder a la comunicación del mismo a las personas implicadas.
La comunicación de los resultados debe ser un proceso al cual las personas que se sometieron al test acudan voluntariamente; se les explica a los sujetos que si realizan el estudio son ellos los que nos tienen que llamar para saber si el resultado ya está disponible y concertar una nueva cita para comentar el mismo; esto es así porque en algunos casos concretos, personas que se habían realizado la prueba convencidas de lo que hacían, valoran la situación de otra manera con el paso de esos meses y deciden que no desean saberlo. Esta situación es totalmente aceptable ya que ante todo debe primar el principio de autonomía de cada persona a conocer o no el resultado sobre su información genética y, sobre todo, porque la utilidad de estos tests en algunos casos no está demostrada así como que las medidas que podamos tomar tampoco tienen una eficacia demostrada.
En cuanto a la forma de comunicar a las personas que se han realizado los estudios lo deseable es realizarlo con una entrevista personalizada que bien se puede producir con cada miembro de la familia por separado o con todos en conjunto. Cada una de las situaciones tiene sus pros y sus contras; la información por separado conlleva una discreción máxima sobre el resultado de una persona en concreto y favorece el que el sujeto nos manifieste todas sus preocupaciones o dudas. En cambio, cuando informamos a toda la familia en grupo, probablemente esta facilidad para preguntar se pierde pero ganamos el que todas las personas reciben la misma información y en el mismo momento, evitando errores de interpretación que puedan conllevar malentendidos en el seno familiar.
Un punto de máxima importancia a lo largo de todo el proceso del consejo genético es la confidencialidad a la hora de recoger los datos en la historia clínica, en el lugar donde se guarda esta información, en el manejo por parte del personal que trabaja con esta información, en la comunicación de resultados y en las personas que tienen acceso a los mismos. Este interés en guardar la confidencialidad de toda la información genética que manejamos implica que a cada persona se le haga conocedor del resultado de su estudio, y nada más que de su estudio, aunque esto pueda implicar el manejo de otras personas.
Las repercusiones psicológicas que el acto del consejo genético puede tener es un punto fundamental en este tipo de consultas y, en algunos casos, va a requerir la intervención de psicólogos especializados en este campo. En este aspecto la valoración psicológica no se debe ofrecer sólo a las personas a las que no conseguimos controlar su distrés ante la situación sino que lo recomendable sería que toda persona que se valora en una Unidad de Consejo Genético tenga una valoración por parte de un profesional en este campo a lo largo de todo el proceso del consejo genético, así como en el seguimiento a largo plazo19,20.
Un punto de interés es el del consejo genético en síndromes oncológicos que afectan a niños (retinoblastoma, poliposis colónicas, etc.). Salvo en estos síndromes en los que el conocimiento de la situación de portador va a conllevar unas medidas preventivas que, en algunos casos, pueden salvar la vida de ese niño, no se debe realizar ningún test genético a los menores de edad. El hecho de que no se conoce muy bien hasta que punto las medidas que podamos tomar son eficaces así como que en la mayoría de los casos no se va a plantear el inicio de medidas preventivas hasta los 20-25 años así como el trastorno psicológico que puede conllevar este tipo de pruebas, apoya el no realizar estos tests hasta que la persona no sea mayor de edad.
El final de todo consejo genético, entre otras cosas, es el poder ofertar a los sujetos en estudio una serie de medidas preventivas que permitan reducir el riesgo de aparición de una enfermedad genética cancerosa o, en el peor de los casos, su detección precoz.
Bases de datos genéticas
La base de datos Mendelian Inheriance in Man (MIM y su versión electrónica Online Mendelian Inheriance in Man, OMIM) es un registro de conocimiento sobre genes humanos y trastornos genéticos. MIM/OMIM nació hace 41 años como un catálogo de fenotipos clínicos mendelianos (1.486 entradas en su primera edición) que ha evolucionado hacia un catálogo de genes mutantes humanos y enfermedades genéticas. Constituye un punto de conexión entre la unidad de expresión clínica y la unidad genética. Esta conexión es particularmente importante para la genética médica porque la definición de la lesión molecular o mutación permite un diagnóstico específico del fenotipo. Este catálogo ha ido en progresión y con fecha de junio de 2008 incluye 18.756 entradas, de las cuales 2.332 se corresponden con fenotipos bien descritos y bases moleculares conocidas (Tabla 1). Estos fenotipos se asocian en su gran mayoría a enfermedades raras. Podemos decir que a lo largo de los últimos 40 o 50 años se ha ido definiendo la biología de la enfermedad y de los modos de enfermar sin precedente en la historia de la medicina y de la humanidad12.
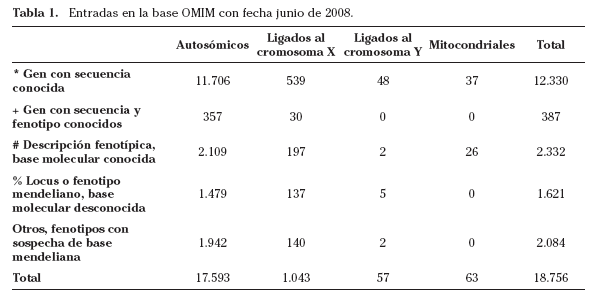
a. Debido a la falta de funcionalidad del enzima que convierte la fenilalanina en tirosina, los niños afectados acumulan ese aminoácido en la sangre, lo que impide el normal desarrollo del cerebro y acaba por originar una grave deficiencia mental.
b. El síndrome de Down es más frecuente, pero, como ocurre de forma espontánea, normalmente no es hereditario.
c. La amniocentesis es causa de aborto en un 1 por ciento de los casos y la toma de células del corion un 2 por ciento.
Bibliografía
1. Strachan T, Read AP. Human Molecular Genetics 3. Oxford: Garland Publishing Bios Scientific Publishers, 2004. [ Links ]
2. VV. AA. Enfermedades raras. JANO, Medicina y Humanidades 2008, nº 1679, pp. 23-44. [ Links ]
3. Antonarakis SE, Beckmann JS. Mendelian disorders deserve more attention. Nature Rev Genet 2006; 7: 277-282. [ Links ]
4. Bickel H, Genard J, Hickmans E. Influence of phenylalanine intake on phenylketonuria. Lancet 1953; 2: 812. [ Links ]
5. Ropers HH. New perspectives for the elucidation of genetic disorders. Am J Hum Genet 2007; 81: 199-207 [ Links ]
6. Pennisi E. The human genome. Science 2001; 291: 1177-1182. [ Links ]
7. Peters JA, Biesecker BB. Genetic counselling and hereditary cancer. Cancer 1997; 80: 576-586. [ Links ]
8. Sobel SK, Cowwan DB. Impact of genetic testing for Huntingtons disease on the family system. Am J Med Genet 2000; 90: 49-59. [ Links ]
9. Baumgardner TL, Reiss AL, Freund LS, Abrams MT. Specification of the neurobehavioral phenotype in males with fragile X syndrome. Pediatrics 1995; 95: 744-752. [ Links ]
10. Triggs-Raine BL, Feigenbaum AS, Natowicz M, Skomorowski MA, Schuster SM, Clarke JT et al. Screening for carriers of Tay-Sachs disease among Ashkenazi Jews. A comparison of DNA-based and enzyme-based tests. N Engl J Med 1990; 323: 6-12. [ Links ]
11. Peltonen L, Perola M, Naukkarinen J, Palotie A. Lessons from studying monogenic disease for common disease. Hum Mol Genet 2006;15: R67-R74. [ Links ]
12. McKusick VA. Mendelian inheritance in man and its online version, OMIM. Am J Hum Genet 2007; 80: 588-604. [ Links ]
13. Korenberg JR, Kawashima H, Pulst SM, Ikeuchi T, Ogasawara N, Yamamoto K et al. Molecular definition of a region of chromosome 21 that causes features of the Down syndrome phenotype. Am J Hum Genet 1990; 47: 236-246. [ Links ]
14. Martínez Frías ML. Frecuencia al nacimiento de niños con síndrome de Down en España: análisis por años y por comunidades autónomas. Efecto del diagnóstico prenatal. Prog Diagn Prenatal 1996; 8: 327-338. [ Links ]
15. OConnor TP, Crystal RG. Genetic medicines: treatment strategies for hereditary disorders. Nature Rev Genet 2006; 7: 261-276. [ Links ]
16. Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill, 1995. [ Links ]
17. Brinkman RR, Dubé M-P, Roulaeu GA, Orr AC, Samuels ME. Human monogenic disorders – a source of novel drug targets. Nature Rev Genet 2006; 7: 249-260. [ Links ]
18. Lemon SJ, Tinley DT, Fusaro RM, Lynch HT. Cancer risk assessment in a hereditary cancer prevention clinic and its first year´s experience. Cancer 1997; 80: 606-613. [ Links ]
19. Croyle RT, Achilles JS, Lerman C. Psychologic aspects of cancer genetic testing. Cancer 1997; 80: 569-575. [ Links ]
20. Kash KM, Lerman C. Psychological, social, and ethical issues in gene testing. Psychooncology. Jimmie Holland. Oxford university Press, 1998. [ Links ]
Direcciones electrónicas
1. GeneReviews; http://www.geneclinics.org/: recurso de información pública sobre genética médica (fenotipo, clasificación y genes de enfermedades genéticas).
2. Human Genome Variation Society; http://www.hgvs.org/mutnomen: portal sobre la nomenclatura de las variaciones de la secuencia del genoma.
3. Nacional Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/: portal de información en biología molecular; NCBI crea bases de datos públicas y desarrolla herramientas de análisis de datos genómicos. Difunde información biomédica e incluye un número amplio de bases de datos como PubMed relativo a bibliografía biomédica, genes, mapas genéticos, proteínas, estructuras moleculares, etc.
4. Online Mendelian Inheritance in Me; http://www.ncbi.nlm.nih.gov/omim: catálogo sobre genes humanos y enfermedades genéticas desarrollado por Victor McKusick en la Universidad Johns Hopkins en Baltimore, EE.UU. desde 1967.
5. The Human Gene Mutation Database HGMDO, http://www.hgmd.cf.ac.uk/: base de datos y registro de mutaciones génicas humanas gestionada en Cardiff, Reino Unido.
6. The Internacional HapMap Projec, http://www.hapmap.org/: soporte electrónico del The International HapMap Project; Mapa de polimorfismos SNPs y haplotipos en cuatro poblaciones humanas, caucasiana de origen europeo, africana y oriental (china y japonesa). Ayuda a los investigadores a encontrar genes asociados a enfermedades humanas y respuesta a fármacos.
7. Portales informativos sobre enfermedades raras y asociaciones de enfermos y familiares, Federación Española de Enfermedades Raras: http://www.enfermedades-raras.org/, European Organisation of Rare Diseases http://www.eurordis.org/, Oficina Norteamaricana de los National Health Institutes http://rarediseases.info.nih.gov/.
![]() Dirección para correspondencia:
Dirección para correspondencia:
Domingo González-Lamuño Leguina
Facultad de Medicina
Universidad de Cantabria-Hospital Universitario Marqués de Valdecilla
Cardenal Herrera Oria s/n
39011 Santander
E-mail pedgld@humv.es

