










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versión impresa ISSN 1137-6627
Anales Sis San Navarra vol.32 no.3 Pamplona sep./dic. 2009
Encefalopatía por hiperamoniemia en el adolescente
Encephalopathy by hyperammonemia in the teenager
B. Zalba1, T. Serrano2, C. Velilla1, J.L. Cabrerizo3, R. Ridruejo1, B. Obón1
1. Unidad de Cuidados Intensivos. Hospital Clínico Universitario. Zaragoza.
2. Servicio de Aparato Digestivo. Hospital Clínico Universitario. Zaragoza.
3. Servicio de Medicina Interna. Hospital Clínico Universitario. Zaragoza.
Dirección para correspondencia
RESUMEN
Fundamento. La hiperamoniemia origina múltiples alteraciones, principalmente en el sistema nervioso central. Si el fallo hepático no es su causa, deberán investigarse otras etiologías menos frecuentes intentando llegar a un diagnóstico definitivo.
Caso clínico. Presentamos el caso de un paciente de 16 años que ingresó por encefalopatía aguda e hiperamoniemia. Tras realización de analítica, TAC cerebral, ecografía y Doppler abdominal, se inició tratamiento empírico de hiperamoniemia secundaria a un error innato del ciclo de la urea. Se trató el edema cerebral y se tomaron medidas para eliminación del amoniaco sin resultado favorable, falleciendo a los cuatro días del ingreso.
Conclusiones. El complejo manejo de la hiperamoniemia y la alta morbi-mortalidad que conlleva requiere un manejo multidisciplinar. La instauración de tratamiento precoz e identificación de la causa son claves para mejorar los resultados.
Palabras clave. Hiperamoniemia. Ciclo de la urea.
ABSTRACT
Background. Hyperammonemia causes several alterations, mainly in the central nervous system. If hepatic failure is not its etiology, other less frequent causes must be investigated in the search for a definitive diagnosis.
Clinical case. We report the case of a 16 year old patient admitted for acute encephalopathy and hyperammonemia. After analysis, brain CT, ultrasound and abdominal Doppler, we began empirical treatment of hyperammoniemia secondary to disorders of the urea cycle. We treated the brain edema and eliminated ammonia but we did not obtain favourable results and the patient died four days later.
Conclusions. The complex management of hyperammonemia and the high morbidity and mortality involved require a multidisciplinary approach. Only early treatment and identification of the hyperammonemia´s etiology can avoid high morbidity and mortality in these patients.
Key words. Hyperammonemia. Urea cycle.
Introducción
La presencia de hiperamoniemia y encefalopatía grave obliga a la búsqueda de un diagnóstico preciso de cara a evitar graves secuelas e incluso la muerte. Diversas enfermedades pueden causar aumento del amoniaco: alteración hepática (hepatitis, isquemia, cirrosis, obstrucción biliar), insuficiencia cardiaca congestiva severa, eritroblastosis fetal, hemorragia digestiva alta, trastornos en el ciclo de la urea, leucemia, pericarditis, síndrome de Reye y algunos fármacos.
Deben establecerse de forma precoz medidas para estabilizar al paciente, eliminar el amoniaco y tratar el edema cerebral1.
Caso clínico
Varón de 16 años con antecedentes personales de adenoidectomía a los 4 años, déficit de factores VII y X (detectado a los 4 años), hiperbilirrubinemia no conjugada (detectada a los 15 años). Tío materno de 37 años con retraso mental fallecido por status convulsivo. Ingresa por cuadro de astenia, hipotensión (100/70 mmHg), anorexia y mareos coincidiendo con descenso de dosis de dexametasona (Dacortin 60 mg/día) prescrita por parálisis facial periférica. Ante la sospecha de insuficiencia suprarrenal se ingresó en planta de hospitalización. Se detectó progresiva tendencia al sueño, ligera desorientación y alucinaciones con hiperbilirrubinemia e hiperamoniemia, por lo que ingresa en la Unidad de Cuidados Intensivos. A su llegada presentó la siguiente exploraración: talla 1,85 m, peso 75 kg, tensión arterial 130/70 mmHg; frecuencia cardiaca 100 lpm; temperatura 36,5o, saturación de O2 100%; consciente pero desorientado; ictérico; resto de exploración normal.
Analítica al ingreso: Hb 16 g/dl (13-17,4), leucocitos 10.800 mil/mm3 (4-11), plaquetas 196.000 mil/mm3 (150-400), fibrinógeno 287 mg/dL (200-450), bilirrubina total 7,1 mgr/dL (0-0,3), bilirrubina directa 0,47 mgr/dL (0,3-1,9), AST 50 UI/L (10-40), ALT 169 UI/L (10-40), GGT 16 UI/L (5-40), amoniaco 424 mgr/dL (15-45), LDH 310 UI/L (210-420). Glucosa 118 mg/dL (69-100), urea 0,17g/L (0,1-0,7), creatinina 0,7 mg/dL (0,5-1), Na 145 mEq/L (132-145), K 3,5 mEq/L (3,1-5,1), Ca 9.1 mEq/L (8,5-10,2), Mg 1,7 mEq/L (1,7-2,2), CO3H 25,78 mEq/L (21-29). Se realizaron las siguientes pruebas de imagen: TAC cerebral sin hallazgos reseñables con buena diferenciación de sustancia blanca y gris. Ecografía abdominal: no signo de trombosis, hígado homogéneo sin alteraciones, riñones, bazo normales. Eco-Doppler abdominal: sin alteraciones en hígado, bazo, vejiga ni riñones. No ascitis, masas ni trombosis. Arteria hepática y vena esplénica normales.
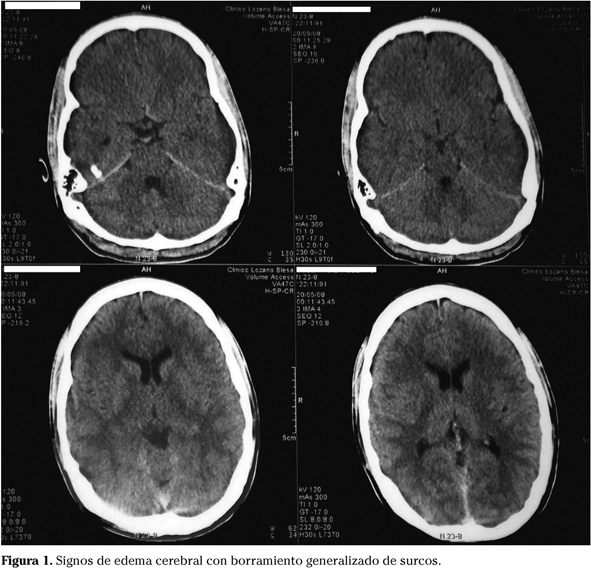
A las 24 h de ingreso cabe destacar elevación del amoniaco hasta 602 mg/dL y actividad de protrombina 44% (75-130) (control de 3 meses antes Act. Pr. 45%). Al día siguiente, descenso paulatino del amoniaco hasta 67 mg/dL con aumento de BT a 10,3 mgr/dL y BD 0,3 mgr/dL y 24 horas después intensa agitación psicomotriz, seguido de coma con GCS 7. Se procede a intubación orotraqueal y se realiza TAC cerebral que es normal y punción lumbar también normal. El amoniaco en este momento era de 575 mg/dL, BT 12,7 mg/dL y BD 0,3 mg/dL. Ocho horas después crisis comicial y amoniaco de 1.723 mg/dL, momento en que iniciamos tratamiento empírico ante la sospecha de un trastorno del metabolismo de la urea, con Benzoato Na 12 g en 30 min y 15 g/24h pc, Carbaglu 1,5 gr/6h IV, L carnitina 3 mg cada 5 h IV, plasmaféresis y hemodiafiltración venovenosa contínua. Doce horas después logramos descenso de BT a 5,4 mg/dL pero el amoniaco se eleva a 4.535 mg/dL, bajando a 4.004 mg/dL 12h más tarde. En este momento el paciente presentaba TAC con signos de edema cerebral (Fig. 1), exploración neurológica y Doppler transcraneal compatibles con muerte encefálica, confirmado por gammagrafía cerebral. Se denegó la necropsia pero sí se realizó biopsia hepática postmorten que mostró hígado con arquitectura conservada, necrosis centrolobulillar de tipo isquémico y cambios sugestivos de patología metabólica. El examen posterior de sangre y orina (en centro de referencia) demostró el déficit de ornitín transcarbamilasa. El estudio de la familia determinó como portadora a la madre y afectos uno de los hermanos y una tía materna.
Discusión
El amoniaco es un producto del metabolismo de los aminoácidos y se elimina en el hígado a través de la síntesis de urea. La fuente principal de los aminoácidos es la dieta y la síntesis «de novo», cuando un aminoácido rebasa las necesidades biosintéticas, éste es destruido y eliminado por transaminación y desaminación oxidativa formando amonio y cetoácidos, utilizados éstos para producir energía en las vías metabólicas produciendo CO2, agua, glucosa, ácidos grasos o cuerpos cetónicos. Si la cantidad de amonio excede la capacidad del ciclo hepático para procesarla se produce la hiperamoniemia originando múltiples alteraciones, principalmente en el sistema nervioso central.
En el caso que nos ocupa se realizaron pruebas diagnósticas orientadas en primer lugar a enfermedades hepáticas como causa del cuadro clínico, más aún existiendo el diagnóstico previo de hiperbilirrubinemia, sin hallar ninguna que lo justificase. Descartadas también otras etiologías de encefalopatía metabólica2 y de formas adquiridas de hiperamoniemia (acidosis tubular distal, tratamiento con valproato, 5FU o anestésicos, shunt portosistémico, derivaciones urinarias, NPT, desnutrición o ingesta de zumos semi-sintéticos) y recordando el antecedente familiar, se llegó a la conclusión de que podría tratarse de un defecto del metabolismo de ciclo de la urea, posiblemente parcial, dada la edad del paciente y su estado asintomático previo. El tratamiento fue iniciado a las 72 horas de ingresar en la Unidad de Cuidados Intensivos (5o día del ingreso en el hospital).
Los trastornos del ciclo de la urea son enfermedades metabólicas hereditarias. Su incidencia es de 1-8.200 nacidos vivos3 y la herencia autonómica recesiva o ligado al X (como en el déficit de ornitin transcarbamilasa)4. Derivan de una anomalía en una de las enzimas que participan en la síntesis de la urea.
Están descritas seis posibles enfermedades según el déficit enzimático implicado: carbamilfosfato sintetasa, ornitina transcarbamilasa, argininosuccinato sintetasa, argininosuccionato liasa, arginasa, n-acetilglutamato sintetasa. La mayoría de los afectados son recién nacidos, aunque los déficits parciales pueden dar síntomas en la infancia o de adultos.
En un estudio de 260 casos el 34% eran neonatos. La edad media de presentación fue 2 años (1 día-53 años) y los síntomas más frecuentes: disminución del nivel de conciencia, status mental alterado, edema cerebral con hiperventilación, déficit de función motora, crisis convulsivas (50%), náuseas, vómitos, diarrea o estreñimiento5.
Pacientes con déficit parcial enzimático como las mujeres portadoras, pueden tener presentaciones atípicas en relación con la ingesta proteica o ante el estrés catabólico: vómitos, retraso en el desarrollo, cefaleas, ataxia, epilepsia o enfermedades psiquiátricas. Las descompensaciones en estos pacientes suelen deberse a cuadros víricos, cirugía, traumatismos, embarazo y uso de corticoides6, pudiendo ser este último el desencadenante en nuestro caso.
El tratamiento se basa en disminuir el amoniaco, sustituir los aminoácidos deficitarios y en última instancia realizar transplante hepático.
Por otra parte, el manejo precoz de la hipertensión endocraneal es esencial, pues los efectos de la hiperamoniemia son muy graves a nivel cerebral1.
Para descender el amoniaco la hemodiálisis es el método más rápido y eficiente y debe realizarse simultáneamente al tratamiento médico7. Se continuará hasta que las cifras de amoniaco queden por debajo de 200 mg/dL. Respecto al tratamiento médico, se utiliza fenilacetato y benzoato de sodio que crean vías alternativas para excretar los precursores del N8, se combinan con glutamina y glicina respectivamente, dando lugar a fenilacetilglutamina e hipurato, ambos solubles y excretados por la orina9,10. Ambas sustancias están combinadas en el preparado Ammonul®, aprobado para uso parenteral por la FDA en 2005. En cuanto a la nutrición es importante restringir el aporte proteico y aportar las calorías necesarias con glucosa y lípidos e iniciar nutrición enteral cuanto antes.
En un reciente estudio sobre 299 pacientes la supervivencia fue del 84% pero el pronóstico neurológico no fue seguido. Algunos estudios muestran que las secuelas neurológicas están en relación con la duración de la hiperamoniemia11.
Todas las medidas citadas deben instaurarse lo antes posible, a pesar de lo cual, como en este caso, no siempre son efectivas de forma inmediata.
Por último, se debe considerar la posibilidad de transplante hepático en algunos candidatos debido a la alta mortalidad y posibles secuelas neurológicas12,13. Debe realizarse una valoración multidisciplinaria y un cuidadoso análisis del riesgo-beneficio. En la serie más extensa hallada la supervivencia fue del 91%, la hiperamoniemia y la dieta restrictiva fueron erradicadas en estos pacientes14. Actualmente también se valora la posibilidad de realizar transplante de hepatocitos como puente a un transplante ortotópico parcial hepático15.
Bibliografía
1. Clay AS, Hainline BE. Hyperammonemia in the ICU. Chest 2007; 132: 1367-1378. [ Links ]
2. Macías MD, López M, Macía C, Fernández A. Hiponatremia aguda grave en un paciente esquizofrénico potomano. An Sist Sanit Navar 2009, 32: 117-120. [ Links ]
3. Vilchez JJ. Enfermedades del sistema nervioso por errores metabólicos congénitos. Medicine 2003; 8: 5548-5557. [ Links ]
4. Brusilow SW, Maestri NE. Urea cycle disorders: diagnosis, pathophysiology and therapy. Adv Pediatr 1996; 43: 127-170. [ Links ]
5. Summar ML, Dobbelaere D, Brusilow S, Lee B. Diagnosis, symptoms, frequency and mortality of 260 patients with urea cycle disorders from a 21-year multicentre study of hyperammonaemia episodes. Acta Pediatr 2008; 97: 1420-1425 [ Links ]
6. Morton GB, Gudmundur J, Margot U. Impact of acute and chronic low-dose glucocorticoids on protein metabolism. J Clin Endocrinol Metab 2007; 92: 3923-3929. [ Links ]
7. Kamoun PP, Rabier D. Valproate-induced inhibition of urea synthesis. Lancet 1987; 1: 48. [ Links ]
8. Summar M. Current strategies for the management of neonatal urea cycle disorders. J Pediatr 2001; 138: S30. [ Links ]
9. Darmaun D, Welch S, Rini A, Sager BK, Almotare A, Haymond MW. Phenylbutyrate-induced glutamine depletion in humans: effect on leucine metabolism. Am J Physiol 1998; 274: E801-807. [ Links ]
10. Green TP, Marchessault RP, Freese DK. Disposition of sodium benzoate in newborn infants with hyperammonemia. J Pediatr 1983; 102: 785-790. [ Links ]
11. Msall M, Batshaw ML, Suss R, Brusilow SW, Mellits ED. Neurologic outcome in children with inborn errors of urea synthesis. Outcome of urea cycle enzymopathies. N Engl J Med 1984; 310: 1500-1505. [ Links ]
12. Saudubray JM, Touati G, Delonlay P, Jouvet P, Narcy C, Laurent J et al. Liver transplantation in urea cycle disorders. Eur J Pediatr 1999; 158 (Suppl 2): 55-59. [ Links ]
13. Whitington PF, Alonso EM, Boyle JT, Molleston JP, Rosenthal P, Emond JC et al. Liver transplantation for the treatment of urea cycle disorders. J Inherit Metab Dis 1998; 21 (Suppl 1): 112-118. [ Links ]
14. Morioka D, Kasahara M, Takada Y, Shirouzu Y, Taira K, Sakamoto S et al. Current role of liver transplantation for the treatment of urea cycle disorders: A review of the worldwide English literature and 13 cases at Kyoto University. Liver Transpl 2005; 11: 1332-1342. [ Links ]
15. Puppi J, Tan N, Mitry RR, Heghes RD, Lehec S, Mieli-Vergani G et al. Hepatocyte transplantation followed by auxiliary liver transplantation--a novel treatment for ornithine transcarbamylase deficiency. Am J Transplant 2008; 8: 452-457. [ Links ]
 Dirección para correspondencia:
Dirección para correspondencia:
Begoña Zalba Etayo
Avda. San Juan Bosco, no 15
Hospital Clínico Universitario
50009 Zaragoza
Tfno. 626807465
E-mail: bzalba2002@yahoo.es
Recepción: 30 de abril de 2009
Aceptación provisional: 27 de mayo de 2009
Aceptación definitiva: 1 de septiembre de 2009

