










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAnales del Sistema Sanitario de Navarra
versión impresa ISSN 1137-6627
Anales Sis San Navarra vol.34 no.3 Pamplona sep./dic. 2011
https://dx.doi.org/10.4321/S1137-66272011000300017
Tumor neuroectodérmico renal primitivo. A propósito de un caso
Primitive neuroectodermal tumour of the kidney. A case report
Dirección para correspondencia
Sr. Director:
Los tumores neuroectodérmicos primitivos de origen renal (rPNET) son neoplasias excepcionales y pertenecen al grupo de tumores neuroectodérmicos periféricos malignos1-3.
Su origen no está claro, aunque se cree que se originan a partir de células migradas desde el tubo neural con capacidad de diferenciación ectodérmica o neuronal variable.
Son tumores extremadamente agresivos, con tendencia a la recurrencia y a la metastatización temprana. La presentación clínica, el patrón histológico y las técnicas de inmuhistoquímica orientan a un diagnóstico que no deja de ser dificultoso debido a su escasa frecuencia y a que en ocasiones ha de verse apoyado en técnicas de citogenética y de análisis molecular.
Esta entidad puede ser confundida con toda la variedad de tumores de células redondeadas de pequeño tamaño. De manera que es importante hacer el diagnóstico diferencial con ellas por sus implicaciones terapéuticas y pronósticas.
Paciente de 29 años de edad que acude a la consulta de urología por presentar dolor en fosa lumbar izquierda, febrícula y microhematuria, de varios meses de evolución.
Como antecedentes personales de interés destaca una obesidad mórbida con un índice de masa corporal (IMC) >35 Kg/m2. No se palparon masas abdominales ni en flancos a la exploración física.
Dentro de las exploraciones complementarias, se objetivó en la analítica unas cifras de creatinina de 1,2 mg/dl. Se realizó una ecografía abdominal en la que se observó la presencia de una lesión quística de unos 13 x 8 centímetros en el riñón izquierdo. Ante estos hallazgos se decidió completar el estudio con una tomografía axial computerizada abdominal (TAC).
El scanner confirmó la presencia de una colección subcapsular sin captación de contraste sugestiva de hematoma (Fig.1).
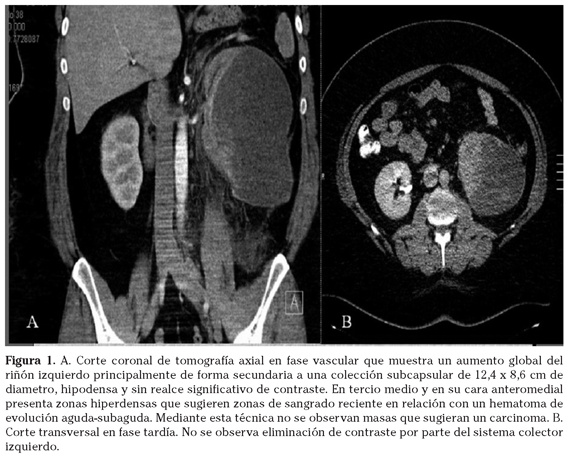
Dicha colección condicionaba una compresión del parenquima y del hilio renal, siendo el riñón no funcionante.
No pudo confirmarse un punto de sangrado activo con la tomografía. Tampoco podía descartarse la existencia de una neoplasia como causa del hematoma.
Por ello se realizó una arteriografía renal observándose permeabilidad de la arteria renal izquierda con un calibre normal. Además se identificó una lesión con neovascularización en el polo inferior, que no desaparecía tras la inyección de 0,01 mg de adrenalina. La imagen resultó ser compatible con un hipernefroma.
Se programó de manera electiva una nefrectomía ampliada laparoscópica izquierda, que tuvo que realizarse con carácter urgente a los pocos días debido a una reagudización del cuadro clínico, y a un incremento del hematoma objetivado en un nuevo scanner.
El diagnóstico anatomopatológico informó de tumor neuroectodérmico primitivo de origen renal (rPNET), de 9 centímetros (Fig. 2).
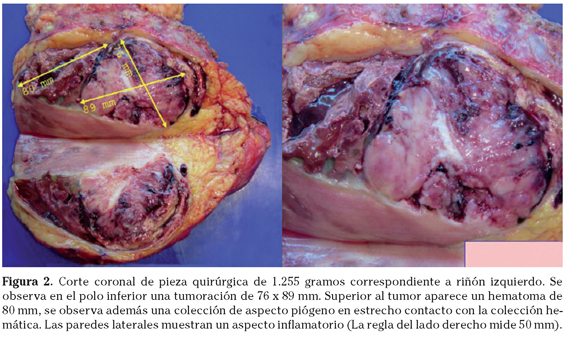
El tumor producía infiltración de la grasa adyacente, de la vena y de la cápsula renal. Los márgenes de resección se encontraron libres de tumor.
El estudio inmunohistoquímico reveló unas células que presentaban positividad para los marcadores vimentina, O13 (CD-99), y WT1, siendo negativos para citoqueratina, desmina, S-100 y CD-34 (Fig. 3).

Tres meses más tarde, se realizó una tomografía emisora de positrones (PET) de control, evidenciando focos hipermetabólicos compatibles con implantes peritoneales. En los segmentos hepáticos VI y VII también se observaron depósitos hipercaptantes sugiriendo enfermedad metastásica.
Bajo control tomográfico se realizó una biopsia de un implante siendo informado de tumor neuroectodérmico periférico.
Debido a la situación actual de progresión de la enfermedad, el paciente inició tratamiento quimioterápico siguiendo el esquema VAC-IE (vincristina, adriamicina, cisplatino, ifosfamida y etopósido), con adecuada respuesta y buena tolerancia actual al mismo.
El riñón de Page supone un cuadro de constricción parenquimatosa renal debido a un hematoma subcapsular o un gran hematoma perirrenal, capaces de alterar la microcirculación intrarrenal, alterando el equilibrio hemodinámico y provocando fenómenos isquémicos que provocan la hiperplasia de las células yuxtaglomerulares, aumentando así la cantidad de renina circulante y elevando las cifras de tensión arterial. Actualmente la causa más frecuente de este tipo de hipertensión hiperreninémica es el hematoma subcapsular, derivado de la biopsia renal en los pacientes después de la realización de un trasplante.
Dado estos datos, el riñón de Page fue un posible diagnóstico que se incluyó en el diagnóstico diferencial de esta patología. La hemorragia retroperitoneal atraumática o Síndrome de Wünderlich fue otra de las opciones que se barajaron como primera impresión diagnóstica. El adenocarcinoma renal seguido del angiomiolipoma, suelen ser las causas más habituales de esta infrecuente entidad, que puede comprometer seriamente la vida del paciente.
Los tumores neuroectodérmicos incluyen a un grupo heterogéneo de entidades patológicas formadas por neoplasias con células redondeadas de pequeño tamaño, tales como el tumor de Wilms, el neuroblastoma, el linfoma, el sarcoma de células claras, el sarcoma de Ewing, la variante de célula pequeña del osteosarcoma, el rabdomiosarcoma, el carcinoma neuroendocrino anaplásico de células pequeñas, el tumor carcinoide y el tumor neuroectodérmico renal primitivo, entre otros1-3.
Se caracterizan por ser uno de los tumores malignos histologicamente más indiferenciados, extremadamente raros y agresivos, y con una elevada tendencia a recurrir localmente y a la diseminación temprana a distancia. Tal es su grado de desdiferenciación y agresividad que en el momento del diagnóstico, el 25-50% de los sujetos presentan metástasis3. Los ganglios linfáticos, el pulmón, el hígado, el hueso y la médula osea, son los órganos de invasión más habituales, condicionando su afectación un peor pronóstico.
La localización renal es muy rara, además presenta un comportamiento más agresivo que los tumores neuroectodérmicos de otras localizaciones y que los tumores renales convencionales. La invasión de la cápsula de Gerota y de la grasa perirrenal suele ser la norma en más de la mitad de los casos.
La clínica no difiere a la de otros tumores renales. La hematuria, el dolor lumbar o abdominal y la masa palpable suelen ser los síntomas más frecuentes. Las pruebas de imagen tampoco aportan características específicas.
Es importante aplicar un repertorio diagnóstico completo, ya que la utilización de una única herramienta puede ser insuficiente para excluir el gran número de diagnósticos diferenciales1.
La microscopía óptica convencional del rPNET muestra una neoformación de estirpe mesenquimal de crecimiento difuso, con células desdiferenciadas redondeadas y de pequeño tamaño, de nucleos ovalados uniformemente teñidos con disminución irregular de la cromatina. En ocasiones estas células adquieren una disposición que tiende a formar rosetas y pseudorrosetas, se conocen con el nombre de rosetas de Hommer-Wright2,3, generalmente escasean en el sarcoma de Ewing mientras que si pueden encontrarse en el caso del neuroblastoma.
Pueden observarse también abundantes zonas de necrosis acompañadas de hemorragia y un gran número de mitosis.
El parénquima renal no tumoral suele mostrar atrofia tubular e infiltración linfocitaria.
El análisis inmuhistoquímico es necesario para este tipo de tumores. La expresión de productos génicos relacionados con las citoquinas inhibitorias de macrófagos (MIC-2), conocidos como CD-99, 12 E7, E2, O13 y HBA71, son sugestivos de PNET aunque no específicos. Actualmente se considera clave la expresión de CD-99, sin embargo no puede utilizarse de manera aislada ya que su positividad es compartida con el sarcoma extraóseo de Ewing.
Se deben utilizar entonces marcadores neuronales para excluir el sarcoma de Ewing.
De esta manera, la reactividad a marcadores neuronales tales como la vimentina, enolasa de neurona específica (NSE), y S-100, no siendo patognomónicos pueden facilitar el diagnóstico6.
Los estudios citogenéticos y las técnicas de análisis molecular quedarían relegados a aquellos casos en los que la combinación de la histopatología e inmunohistoquímica resultasen insuficientes para establecer un juicio clínico.
La técnica de hibridación fluorescente in situ (FISH) ha demostrado una traslocación cromosómica, t (11:22) (q 24; q12)3-5,7.
Esta mutación corresponde al locus específico EWS/R1 (región 1 del punto de rotura del sarcoma de Ewing) (22q12). La traslocación lleva a la fusión del gen FLI-1 ubicado en el cromosoma 11, produciendo trasncripción de información quimérica.
Mediante la reacción en cadena de la polimerasa (PCR) se ha confirmado que el transcripto de fusión EWS/FLI-1 está presente en el 85% de estos tumores.
No obstante no es la única mutación descrita, ya que se han encontrado otras variantes aunque con menor frecuencia.
Autores como Kumar et al. refieren técnicas que permiten describir con seguridad la fusión EWS/FLI-1 a partir de material fijado, pero estas técnicas aun no están generalizadas3,7.
Debido a la semejanza biológica con el sarcoma de Ewing, el PNET puede ser manejado con el mismo protocolo quimioterápico, EFT-2001 (Ewing´s family tumors)2. Consiste en la administración intensiva de un ciclo que suele alternar una media de dos fármacos quimioterápicos con una duración semanal. Ciclofosfamida, Actinomicina, Vincristina, Doxorubicina, Ifosfamida y Etoposido suelen ser las drogas empleadas. El ciclo se repite cada tres semanas hasta cumplir un total de 49 semanas, con una media de 17 ciclos.
Otros protocolos como el RCT II (round cell tumor), VAC (vincristina, adriamicina, cisplatino) y VA (vincristina, adriamicina), también han sido en ocasiones utilizados.
El consenso es difícil debido a la escasez de estudios randomizados publicados sobre la efectividad de los diferentes regímenes de quimioterapia.
La radioterapia adquiere su papel en aquellos casos en los que la cirugía no es posible y para un adecuado control local de la enfermedad, pacientes con márgenes positivos o con afectación de la fascia de Gerota. La dosis de radiación empleada varía entre 5040-6000 rads.
En cuanto al pronóstico, con tratamiento agresivo multimodal combinando cirugía, quimioterapia y radioterapia, la tasa libre de enfermedad a los 5 años es de un 45-55%1-3. Cuando la enfermedad está diseminada al diagnóstico, la supervivencia media es de 2 años.
R. Algarra, J. Barba, L.M. Romero, F.J. Queipo, D. Rosell, F.J. Pardo
Clínica Universidad de Navarra. Pamplona. Navarra. España.
Bibliografía
1. Cabrera-Meirás F, Duque Ruiz G, Martínez-Silva VM, Leiva-Galvis O. Primitive neuroectodermal tumor of the kidney: Case report. Actas Urol Esp 2010; 34: 730-732. [ Links ]
2. Yuvaraja B, Thyavihally YB, Tongaonkar HB, Gupta S, Kurkure PA, Amare P et al. Primitive neuroectodermal tumor of the kidney: a single institute series of 16 patients. Urology 2008; 71: 292-296. [ Links ]
3. Mallén Mateo E, Sancho Serrano C, Pascual Regueiro D, García de Jalón Martínez A, Gil Sanz MJ, Rioja Sanz LA. Primitive neuroectodermal tumor. Ewing´s Sarcoma. Actas Urol Esp 2005; 29: 311-313. [ Links ]
4. Cuesta Alcalá JA, Solchaga Martínez A, Caballero Martínez MA, Gómez Dorronsoro M, Pascual Piedrola I, Ripa Saldías L et al. Tumor neuroectodérmico primitivo renal (PNET): 26 casos. Estado actual de su diagnóstico y tratamiento. Arch Esp Urol 2001; 54: 1081-1093. [ Links ]
5. Kumar S, Pack S, Kumar D, Walker R, Quezado M, Zhuang Z et al. Detection of EWS-FLI-1 fusion in Ewing´s sarcoma/peripheral primitive neuroectodermal tumor by fluorescence in situ hybridization using formaling-fixed paraffin -embedded tissue. Hum Pathol 1999; 30: 324-330. [ Links ]
6. Gonzalo Valero F, Arturo Escalona L, Rodrigo Leyton N. Tumor neuroectodérmico primitivo de riñón. Caso clínico. Rev Méd Chile 2001; 2: 191-195. [ Links ]
7. Kumar R, Gautam U, Srinivasan R, Lal A, Sharma U, Kumar S et al. Primary Ewing´s sarcoma/primitive neuroectodermal tumor of the kidney: Report of a case diagnosed by fine needle aspiration cytology and confirmed by inmunocytochemistry an RT-PCR along with review of literature. Diagnostic cytopathology 2011 May 4 DOI 10.1002/dc.21717 (Epub ahead of print). [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Rubén Algarra Navarro
Departamento de Urología
Clínica Universidad de Navarra
Avda. Pío XII, 36.
31008 Pamplona. Navarra. España.
E-mail: ralgarra@unav.es
Recepción: 14 de julio de 2011
Aceptación provisional: 1 de agosto de 2011
Aceptación definitiva: 24 de agosto de 2011

