










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
El síndrome de Churg-Strauss (SCS) se describió como una forma de vasculitis necrotizante diseminada con granulomas extravasculares que ocurre en pacientes con asma y eosinofilia. En 2012, tras la revisión de la nomenclatura de las vasculitis, se denominó granulomatosis eosinofílica con poliangeítis (GEP)1. La GEP es una vasculitis necrotizante de vasos de pequeño y mediano calibre con una baja incidencia, 0,11-2,66 nuevos casos/106habitantes/año, y una prevalencia de 10-14 casos/106habitantes. Puede afectar a diversos órganos y tiene buen pronóstico, aunque puede tener una evolución mortal, especialmente si se produce afectación cardíaca2.
El objetivo es presentar un caso de evolución mortal de GEP no diagnosticada, en un paciente que previamente había acudido a urgencias hospitalarias, y donde la autopsia permitió caracterizar y diagnosticar el cuadro con afectación multiorgánica y especialmente cardíaca.
Caso clínico
Se trata de un varón de 38 años, de nacionalidad española, desplazado de su zona de residencia, con antecedentes de asma y consumo de cannabis. Acude a urgencias de hospital comarcal por contusión en hemitórax derecho después de una caída por cuadro de un mes de evolución con debilidad progresiva, sin fiebre, con diarreas, vómitos y dolor abdominal asociado a pérdida ponderal de unos 10 kg. En dicha asistencia las constantes vitales eran de 37,3ºC, frecuencia cardíaca 118 lpm, tensión arterial sistólica de 105 mmHg y diastólica de 68 mmHg. La exploración física en el hospital fue normal a excepción de la palidez cutánea y la impresión de palpación de borde hepático de dos traveses de dedo. Se realizó radiología de abdomen sin hallazgos y una analítica que mostró elevación de proteína C reactiva (112,6 mg/L), leucocitosis (23,56 x 103/mm3) de predominio eosinofílico (19%, recuento 4,48 x 103/mm3), creatinina sérica de 1,29 mg/dl y elevación leve de enzimas hepáticas (AST/GOT: 69 U/L, ALT/GPT: 75 U/L). No se realizaron otras pruebas complementarias. Fue dado de alta en urgencias con la orientación diagnóstica de parasitosis intestinal, tratado con metronidazol y ciprofloxacino, y se recomendó control por el médico de su zona para completar estudio (coprocultivo) y seguimiento. A los 12 días, tras continuar con malestar general, anorexia y astenia, presenta parada cardiorrespiratoria no recuperada y fallece.
La autopsia judicial reveló un cadáver con normopeso (IMC: 23,4 kg/m2), lesiones secundarias a maniobras de reanimación cardiopulmonar, edema y congestión pulmonar bilateral (peso de ambos pulmones: 2.011 g), pericarditis y derrame pericárdico de aspecto purulento, cardiomegalia (700 g, en vez del peso esperado de 344 g), necrosis miocárdica de ventrículo izquierdo y músculos papilares, hepatoesplenomegalia (hígado: 2.318 g, bazo: 306 g), focos de necrosis en parénquima hepático y esplénico (Fig. 1) y aspecto isquémico corticomedular renal bilateral. Provisionalmente se orientó como una muerte debida a miocarditis a la espera de los resultados de las pruebas complementarias.

El estudio toxicológico fue negativo y el estudio histopatológico de las vísceras (cerebro, pulmones, corazón, hígado, bazo, riñones e intestinosFig. 2) se informó como vasculitis necrotizante con granulomas extravasculares e hipereosinofilia diseminada afectando a pulmón, corazón, estómago, intestino, hígado, riñones y bazo; infarto cardíaco masivo en evolución con trombosis intracavitaria e infartos agudos hepáticos y esplénicos extensos secundarios al proceso anterior. Además se halló hipertrofia cardíaca con dilatación biventricular, focos dispersos de necrosis cerebral probablemente de origen embólico, cambios de remodelación de la vía aérea compatibles con asma bronquial e infiltrados neumónicos con focos de reorganización. Se concluyó que la muerte fue causada por la necrosis miocárdica masiva debida a GEP.
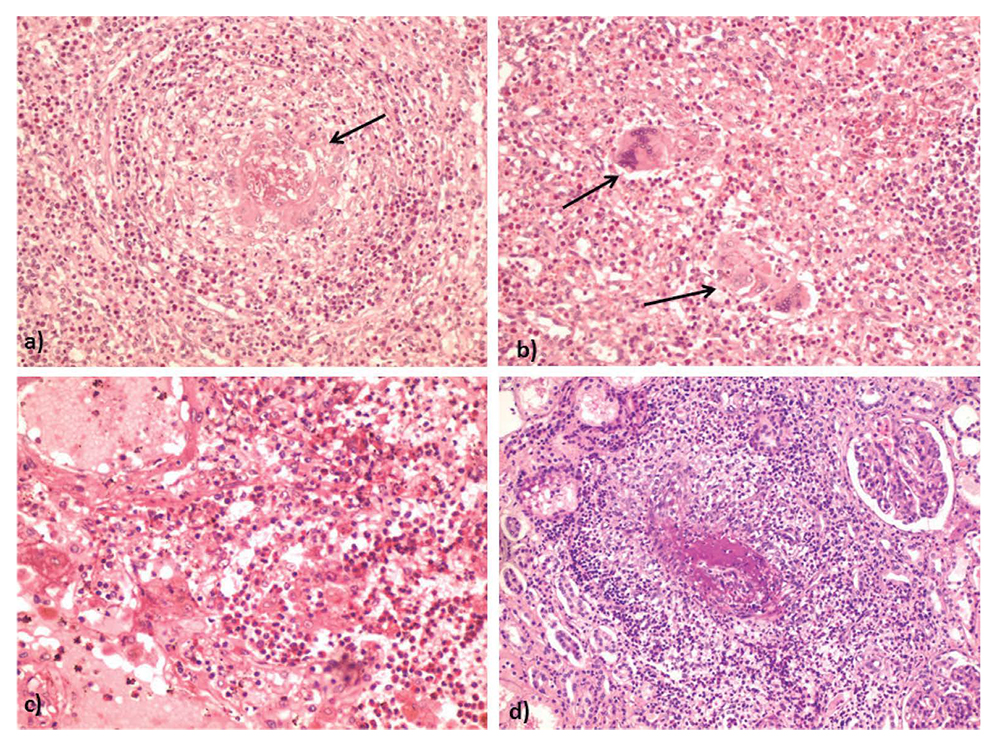
Figura 2. a) Bazo. Granuloma extravascular con necrosis central (flecha); b) Parénquima esplénico con células gigantes multinucleadas (flechas); c) Parénquima pulmonar con edema e infiltrado inflamatorio intraalveolar con predominio de eosinófilos. d) Pared intestinal. Necrosis fibrinoide e infiltrado inflamatorio trasmural en la pared de un vaso de pequeño calibre en la capa submucosa. Hematoxilina-eosina 20x.
Discusión
El caso presentado de evolución mortal y diagnósticopost mortemde una GEP es sumamente infrecuente. En una serie de 3.980 autopsias clínicas de adultos, se identificaron 42 casos de vasculitis sistémica (1,1%), 18 de los cuales fueron vasculitis necrotizantes, con solo dos casos de GEP (4,8%)3.
Los antecedentes clínicos del fallecido, asma e hipereosinofilia periférica del 19% constatados en urgencias, junto con los hallazgospost mortem, permitieron el diagnóstico del cuadro. El estudio histopatológicopost mortemdemostró la tríada característica de este síndrome: infiltración eosinofílica en todos los tejidos, vasculitis necrotizante y granulomas extravasculares. La presencia de estos tres hallazgos es frecuente en casos autópsicos como el nuestro, pero la identificación de granulomas extravasculares (la lesión más específica del GEP) es menos frecuente en los casos clínicos debido a la instauración de la terapia con glucocorticoides y/o al pequeño tamaño de las biopsias1. Desde un punto de vista clínico, el diagnóstico requiere la presencia de cuatro o más de los siguientes criterios propuestos por elAmerican College of Rheumatology: asma, eosinofilia >10%, neuropatía, infiltrados pulmonares, anormalidades en los senos paranasales e infiltración eosinofílica extravascular en la biopsia4. Excepto las anormalidades de los senos paranasales y la neuropatía, el resto de criterios estaban presentes en el caso.
Su presentación clínica es heterogénea y, tradicionalmente, la GEP evoluciona en tres fases, desde una fase prodrómica representada por asma y rinosinusitis, siguiendo con una fase eosinofílica marcada por la eosinofilia periférica y afectación de los órganos, y hasta una fase vasculítica con manifestaciones clínicas debidas a la vasculitis de vasos de pequeño calibre1. En el caso descrito se describen hallazgos propios de la fase eosinofílica con afectación de múltiples órganos (especialmente a nivel cardíaco, pulmonar y gastrointestinal) y de la fase vasculítica. Esta se caracteriza por síntomas constitucionales como los recogidos tanto en la asistencia en urgencias hospitalarias como en el momento de la muerte (febrícula, pérdida de peso de 10 kg, debilidad progresiva, anorexia y astenia), y se confirma histopatológicamente por la presencia de vasculitis necrotizante afectando a múltiples órganos.
A pesar de tratarse de una muerte inesperada por un proceso no diagnosticado previamente, no puede considerarse una muerte súbita, como en el caso descrito por Val Bernal y col en un varón de 49 años sin sintomatología previa5. Como hemos explicado, se trataba de un cuadro clínicamente evolucionado que se correspondía con las fases eosinofílica y vasculítica.
Aunque su pronóstico es habitualmente bueno con un 91% de pacientes que alcanzan la remisión, un 25% de ellos sufren recaídas y un 11% fallecen2. En general la GEP ha sido considerada como una vasculitis sistémica con una mortalidad menor comparada con otras vasculitis y con una tasa de remisión similar a la granulomatosis con poliangeítis y superior a la de la poliangeítis microscópica. Sin embargo, cuando no se trata su mortalidad es del 50% a los tres meses, similar a la granulomatosis con poliangeítis. El tratamiento con glucocorticoides es la piedra angular del tratamiento1.
El Grupo Francés de Estudio de la Vasculitis6identificó cinco factores de mal pronóstico: aumento de creatinina sérica (>1,58 mg/dL), proteinuria (>1 g/día), miocardiopatía, afectación gastrointestinal y afectación del sistema nervioso central. En nuestro caso la creatinina estaba al límite aunque en valores normales, y había afectación cardíaca y gastrointestinal.
La afectación sistemática del corazón ocurre en el 27-47% de los casos y representa la principal causa de fallecimiento precoz. Como en otras miocardiopatías inflamatorias causadas por enfermedades autoinmunitarias sistémicas, su temprano diagnóstico y tratamiento son cruciales para el pronóstico7. A nivel cardiaco, se trataba de un cuadro complejo que traducía también un estado evolutivo avanzado de la enfermedad. Además de extensas zonas de necrosis subaguda, se identificaron amplias zonas confluyentes de necrosis de coagulación sin una distribución coronaria determinada y que estaban en relación con la vasculitis necrotizante y fenómenos trombóticos de las arterias coronarias intramiocárdicas, responsables de la isquemia aguda. También se detectó miocardiopatía inflamatoria que se extendía a la grasa epicárdica y al endocardio, y una pericarditis, manifestación frecuente de la enfermedad.
Los datos clínicos durante la asistencia en urgencias hospitalarias se interpretaron como una infección parasitaria intestinal y se pautó tratamiento con antibióticos. Quizás la asociación de asma e hipereosinofilia podía haber orientado hacia un cuadro de vasculitis sistémica, aunque las vasculitis necrotizantes sistémicas requieren un alto grado de sospecha diagnóstica. Son entidades en las que el diagnóstico de certeza plantea una gran dificultad, debido a que las manifestaciones clínicas son heterogéneas e inespecíficas, aspectos que influyen en el retraso diagnóstico, que a veces solo se obtiene tras el estudiopost mortem3. La enfermedad debe ser diferenciada de la poliangeítis y otros síndromes hipereosinofílicos incluyendo infecciones parasitarias (como ocurrió en nuestro caso) o fúngicas, neumonía eosinofílica (síndrome de Loeffler), vasculitis por medicamentos y granulomatosis de Wegener5. Aunque la GEP pertenece al espectro de las vasculitis asociadas a anticuerpos anticitoplasma de neutrófilos (ANCA), se diferencia de la granulomatosis con poliangeítis (Wegener) y de la poliangeítis por su asociación con asma severo y la presencia de eosinófilos en sangre y tejidos. La negatividad de ANCA no descarta el diagnóstico de la enfermedad8.
El caso presentado destaca por la afectación de múltiples órganos, confirmándose la afectación cardíaca como causa principal de fallecimiento en estos pacientes, por lo que se debe realizar un tratamiento precoz para evitar su evolución fatal.
La baja incidencia y la potencial gravedad de la GEP, proceso que requiere de una alta sospecha para plantear un diagnóstico diferencial ante situaciones de hipereosinofilia en pacientes asmáticos, y la necesidad de instaurar un tratamiento adecuado, hacen relevante la difusión de este caso clínico.

