










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkInternational Microbiology
versión impresa ISSN 1139-6709
INT. MICROBIOL. vol.7 no.3 sep. 2004
| RESEARCH REVIEW | |||
|
| |||
| Hijacking of eukaryotic functions by intracellular bacterial pathogens
Summary. Intracellular bacterial pathogens have evolved as a group of microorganisms endowed with weapons to hijack many biological processes of eukaryotic cells. This review discusses how these pathogens perturb diverse host cell functions, such as cytoskeleton dynamics and organelle vesicular trafficking. Alteration of the cytoskeleton is discussed in the context of the bacterial entry process (invasion), which occurs either by activation of membrane-located host receptors ("zipper" mechanism) or by injection of bacterial proteins into the host cell cytosol ("trigger" mechanism). In addition, the two major types of intracellular lifestyles, cytosolic versus intravacuolar (phagosomal), which are the consequence of alterations in the phagosome-lysosome maturation route, are compared. Specific examples illustrating known mechanisms of mimicry or hijacking of the host target are provided. Finally, recent advances in phagosome proteomics and genome expression in intracellular bacteria are described. These new technologies are yielding valuable clues as to how these specialized bacterial pathogens manipulate the mammalian host cell. [Int Microbiol 2004; 7(3):181-191] Key words: bacterial pathogens · hijacking of functions · cell invasion · vacuoles · cytoskeleton · vesicular trafficking | ||
| |||
| Secuestro de funciones eucarióticas por patógenos intracelulares bacterianos Resumen. Los patógenos bacterianos intracelulares han evolucionado como un grupo de microorganismos altamente especializados en el secuestro de funciones propias de células eucariotas. Esta revisión discute cómo esos patógenos alteran diversas funciones de la célula hospedadora, tales como la dinámica del citoesqueleto y el tráfico vesicular entre orgánulos. Se describe la alteración del citoesqueleto durante el proceso de entrada (invasión) de la bacteria. Este proceso puede desencadenarse bien por la activación de receptores de la membrana de la célula hospedadora (mecanismo de tipo "cremallera") o por la inyección de proteínas bacterianas en el citosol de la célula hospedaora (mecanismo de tipo "activador"). Se comparan también los dos tipos principales de vida intracelular de estos patógenos: en el citosol o en el interior de vacuolas (vida fagosómica). Ambos son consecuencia de cambios de la ruta clásica de fusión fagosoma-lisosoma. Se aportan algunos ejemplos representativos en los que se conoce el mecanismo de mimetismo o de secuestro de funciones eucarióticas. Por último, se mencionan los avances más recientes en la proteómica del fagosoma y en la expresión de genomas en bacterias intracelulares. Estos nuevos enfoques aportan información valiosa sobre cómo estas bacterias patógenas especializadas manipulan la célula huésped de mamífero. [Int Microbiol 2004; 7(3):181-191] Palabras clave: patógenos bacterianos · secuestro de funciones · invasión celular · vacuolas · citoesqueleto · tráfico vesicular | Sequestro de funções eucarióticas por patógenos bacterianos intracelulares Resumo. Os patógenos bacterianos intracelulares tem evoluido como um grupo de microrganismos altamente especializados no seqüestro de funções próprias de células eucarióticas. Esta revisão discute como eses patógenos alteram diversas funções da célula hospedeira, tais como a dinâmica do citoesqueleto e o tráfico vesicular entre organelas. Descreve-se a alteração do citoesqueleto durante o processo de entrada (invasão) da bactéria. Esse processo pode desencadear-se seja pela ativação de receptores da membrana da célula hospedeira (mecanismo do tipo "cremalheira"), ou injeção de proteínas bacterianas no citosol da célula hospedeira (mecanismo do tipo "ativador"). Também foram comparados os tipos principais de vida intracelular destes patógenos: no citosol e no interior de vacúolos (vida fagosómica). Ambos são decorrentes de mudanças da rota clássica de fusão fagossoma-lisossoma. Alguns exemplos representativos foram tratados, nos quais são conhecidos o mecanismo de mimetismo ou de seqüestro de funções eucarióticas. Por último, foram mencionados os avanços mais recentes na proteómica do fagosoma e na expressão de genomas em bactérias intracelulares. Estes novos enfoques trazem informação valiosa sobre a maneira que estas bactérias patogênicas especializadas manipulam a célula hospedeira de mamíferos. [Int Microbiol 2004; 7(3):181-191] Palavras chave: patógenos bacterianos · sequestro de funções · invasão celular · vacúolos · citoesqueleto · tráfico vesicular |
Introduction
Infectious diseases caused by bacterial pathogens are one of the leading causes of morbidity and mortality worldwide. Among the different types of known bacterial pathogens, several have evolved to parasitize the intracellular eukaryotic niche. Researchers interested in the biology of these pathogens have often addressed two basic phenomena: the mode of entry and the intracellular lifestyle of the pathogen within the infected cell. Bacterial uptake has been intensively analyzed in non-phagocytic cells, also known as "non-professional phagocytes". Examples are the epithelial and endothelial cells that form the natural cellular defense barriers. Non-phagocytic cells normally do not ingest microbes or other large particles, and therefore have proved to be excellent models for characterizing events entirely driven by the pathogen. This model of infection has contributed to the differentiation of two mechanisms of pathogen-mediated entry: the binding of bacterial adhesins to host cell receptors and the delivery of bacterial effector proteins into the host cell cytosol. Intracellular bacterial pathogens have also developed a variety of lifestyles within the infected host. A basic distinction consists of whether the pathogen gains access to the host cell cytosol or, in contrast, remains enclosed in a membrane-bound vacuole (phagosome).
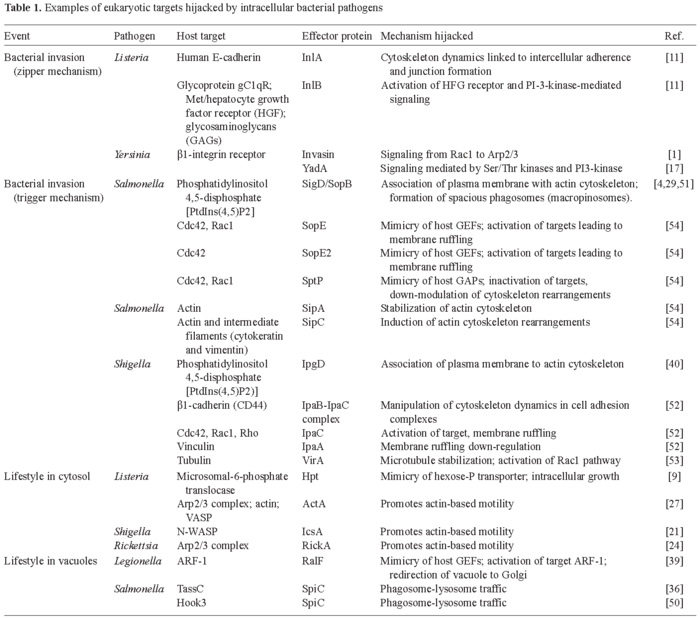
In this review, we summarize how intracellular bacterial pathogens engage specific host cell functions related to cytoskeleton dynamics and host vesicular trafficking in order to promote uptake and ensure intracellular survival and proliferation. Representative examples are listed in Table 1. Due to space limitations, we have based this review on the most recent publications on these broad topics. We therefore apologize to those scientists whose work has not been cited or properly discussed. Related subjects, such as bacterial attachment to host cell surfaces and anti-phagocytosis, are not covered. Excellent recent reviews addressing these topics are recommended [10,27,28,41].
Entry of bacterial pathogens into "professional" phagocytic cells
Phagocytosis of bacteria by specialized "professional" phagocytes is part of the innate immune response of the host. In response, many intracellular bacterial pathogens have evolved to survive and even proliferate within macrophages, neutrophils and dendritic cells. Examples include Brucella spp., Mycobacterium spp., Legionella spp., and Salmonella enterica. The study of the bacterial entry mechanism in phagocytic cells, however, is often hampered by the fact that these pathogens may use their own invasion machinery, leading to an infection process substantially distinct from that of the uptake of opsonized bacteria. The activation state of the phagocyte by immunomodulatory molecules is another relevant factor affecting entry and/or survival of the intracellular pathogen. Based on these considerations, numerous studies have instead focused on comparing receptors and signaling cascades involved in the uptake of opsonized or killed bacteria versus those associated with phagocytosis mediated by the pathogen. Many intracellular pathogens interfere with macrophage signaling in order to prevent killing by these specialized cells [45].
Brucella spp. are intracellular pathogens that survive within macrophages and monocytes. When opsonized, the bacterium is ingested via complement and Fc receptors whereas non-opsonized bacteria use lectin or fibronectin receptors in addition to other, yet undefined receptors [23]. Depending on the route of entry, the fate of intracellular bacteria varies significantly, and opsonized Brucella, for example, are destroyed efficiently. The uptake of Mycobacterium spp. by phagocytes has been intensively studied since these cell types are the preferred targets of this successful pathogen. The heterodimeric host surface receptor C11b/CD18 of the integrin superfamily, known as complement-receptor 3 (CR-3), mediates uptake of opsonized and non-opsonized mycobacteria. Interestingly, CR-3 is targeted by other intracellular pathogens, such as Coxiella burnetii, in order to avoid phagocytosis. This strategy is based on ensuring a spatial location of CR-3 outside the pseudopod extensions. In the case of Salmonella enterica, a correlation exists between the route of entry into phagocytes and the response of the infected cell. Thus, wild-type virulent S. enterica triggers apoptosis in cultured macrophages, an effect that is not observed with a serum-opsonized non-invasive mutant. Unlike the phagocytosis of wild-type bacteria, entry of the opsonized S. enterica mutant is dependent on host tyrosine kinases and phosphatidyl-inositol-3-kinase (PI3-K).
The invasion of non-phagocytic eukaryotic cells
Intracellular bacterial pathogens gain access to non-phagocytic eukaryotic cells via two mechanisms, which were initially differentiated according to morphological criteria. In the first mechanism, "zipper" or "receptor-mediated entry", the invading bacteria are tightly bound to the host cell membrane. The progressive sliding of the bacterium on the host cell membrane ends with the complete inclusion of the invading bacteria. Minor mobilization of cytoskeletal proteins is needed for this type of pathogen-mediated phagocytosis, which is initiated by specific contacts between bacterial ligands (adhesins) and host cell surface receptors (Fig. 1). Unlike the zipper mechanism, the "trigger mechanism" involves dramatic cytoskeletal rearrangements. These alterations consist of membrane extensions in the form of filopodia and lamelopodia at the site of contact of the bacteria with the host eukaryotic cell surface. This activity is known as "membrane ruffling" and depends on the activation of small guanosine triphosphatases (GTPases) of the Rho subfamily (Rho, Rac1 and Cdc42) (Fig. 1). An important mechanistic difference between the zipper and trigger modes of entry is that, whereas the former is promoted from "outside" through activation of host cell receptors, the latter is triggered from "inside" via the action of pathogen-effector proteins delivered by specialized protein secretion systems (Fig. 1).

The zipper invasion mechanism. In this type of entry, a bacterial adhesin binds to a host cell surface receptor involved in cell-to-cell adhesion and/or activation of regulatory proteins that modulate cytoskeleton dynamics. These proteins are often connected to signaling cascades triggered by tyrosine phosphorylation and leading, among other effects, to actin rearrangement and membrane reorganization. The cases more extensively studied are those of Listeria, Yersinia and Helicobacter.
Entry of Listeria monocytogenes into non-phagocytic cells is mediated by at least two proteins, internalin A (InlA) and internalin B (InlB) [11]. InlA is covalently linked to the peptidoglycan by a LPTTG motif located near the C-terminal end. InlA also harbors several leucine-rich repeats (LRRs) in its N-terminal half, probably involved in protein-protein interactions. The host cell surface receptor for InlA is human E-cadherin [11], which is required for optimal intercellular adherence and adherence junction formation. Actin remodeling resulting from binding of InlA to E-cadherin is promoted by α and β-catenins, molecules that normally link the receptor to the cytoskeleton fibers present in the adherence junction [11]. Therefore, Listeria hijacks a previously existing link between a surface receptor and the actin cytoskeleton. Listeria exploits other host signaling pathways by means of InlB. This bacterial surface protein anchors to the cell wall via an interaction between conserved Gly-Trp (GW) modules and lipoteichoic acids. InlB, like InlA, harbors LRR repeats in its N-terminal region and interacts with at least three host surface molecules: the glycoprotein gC1qR (receptor for C1q, the first component of the complement cascade); the tyrosine-kinase receptor Met, also known as hepatocyte growth factor receptor (HGF-R); and glycosaminoglycans (GAGs) [11]. Interaction of InlB with Met (HGF-R) mimics physiological stimulation of the receptor, leading to a cascade of events, including activation of PI-3-kinase, release of phosphatidyl-inositol-3,4,5 triphosphate (PIP3), and activation of Rho GTPases, that cause actin rearrangements [11].
Studies using tissue culture cells have shown that InlB is essential for Listeria uptake by most non-phagocytic cell types, such as hepatocytes, endothelial cells, fibroblasts and certain epithelial cell lines. In contrast, InlA-mediated invasion is apparently restricted to cells expressing E-cadherin, such as those of the intestinal epithelium. Importantly, mutants lacking both InlA and InlB retain, albeit at lower rates, the capacity to enter cultured non-phagocytic cells, suggesting that other adhesins may also promote Listeria entry. Candidates for these putative adhesins are the internalin-like proteins containing LRRs encoded in the genome of this pathogen. In addition, it is known that ActA, a Listeria protein required for actin-tail formation and intracellular cytosolic movement, can also mediate Listeria uptake by epithelial cells.
Yersinia pseudotuberculosis and Y. enterocolitica possess two proteins, invasin and YadA, that promote adhesion to and uptake by epithelial cells. These bacterial adhesins engage a subset of the β1-integrin host receptor family of proteins, a process that is thought to be crucial in the invasion of M cells by these pathogens during the initial steps of infection. Yersinia invasion directed by either invasin or YadA relies on tyrosine and Ser/Thr protein kinases as well as activation of PI3-K [17]. Other host targets hijacked during the entry process are Rac1 and the Arp2/3 complex, which has been implicated in actin recruitment [1]. Synthesis of Inv and YadA adhesin is favored under different environment conditions [17], suggesting that each adhesin is used at a different stage during the infection process. Host molecules activated in response to the adhesion of Y. enterocolitica include the nuclear factor NF-κβ, mitogen-activated protein kinase (MAPK) p38 and the c-Jun N-terminal protein kinase (JNK) [25]. Increased levels of the pro-inflammatory chemokine interleukin-8 (IL-8) have also been reported. A third Yersinia adhesin, Ail, mediates bacterial entry into epithelial cells, although much less is known about its mode of action.
An increasing number of reports suggest that Helicobacter pylori is able to invade non-phagocytic cells. Efficient infection of cultured epithelial cells seems to be restricted to certain H. pylori strains [35]. In all cases, however, invasion of H. pylori seems to involve a typical zipper-like entry process. Both PI3-K and protein-kinase-C (PKC) are required for bacterial uptake and induction of cytoskeletal rearrangements [35]. Invasion of epithelial cells by H. pylori may constitute one of the evasion strategies used by this pathogen to circumvent the host immune response and persist in the human stomach. Bacterial and host factors involved in the entry mechanism are currently unknown.
The trigger invasion mechanism. Bacterial effector proteins that are "injected" into the host cytosol promote this mechanism of invasion. The effector proteins target key host regulatory proteins leading to alteration of signaling cascades that control cytoskeleton dynamics, release of immunomodulatory molecules and host cell survival/death. Delivery of bacterial effectors is carried out by sophisticated secretion machineries, the type III secretion systems (TTSS), consisting of supramolecular protein complexes spanning the cytoplasmic and outer bacterial membranes. TTSS have two well-differentiated parts: a basal body connected to a needle-like structure that spans outwards [10,20]. The mechanism of injection is thought to proceed in a sequential manner: (i) secretion of "translocator" proteins, (ii) their insertion into the host cytoplasmic membrane to form a "pore", and (iii) contact by the tip of the needle and subsequent injection of effector proteins into the host cytosol.
In the discussion that follows, the common and distinct features of the trigger-type entry mechanisms of Salmonella and Shigella will be described. These pathogens induce dramatic cytoskeletal rearrangements resulting in membrane ruffling, macropinocytosis and bacterial entry. Some of the effector proteins delivered by these pathogens mimic guanine-nucleotide-exchange factors (GEFs) or GTPase-activating proteins (GAPs) of members of the Rho subfamily of small GTPases (Cdc42, Rac1 and Rho)[27,54].
Salmonella uses the TTSS encoded in the Salmonella-pathogenicity island 1 (SPI-1) to invade non-phagocytic cells. SigD/SopB is a protein secreted by the SPI-1 TTSS that has phosphatidyl-inositol phosphatase activity. SigD/SopB induces rapid disappearance of phosphatidyl-inositol-4,5-diphosphate PtIns(4,5)P2 from invaginating regions of the cytoplasmic membrane, causing loosening of membrane attachments to the cortical actin cytoskeleton [4,51]. SigD/SopB has recently been implicated in formation of spacious phagosomes (macropinosomes) [29]. Other SPI-1 secreted effectors, SopE and SopE2, activate either Cdc42 and Rac1 (SopE) or Cdc42 alone (SopE2), acting as GEFs of these GTPases [54]. The injection of either SopE or SopE2 is sufficient to cause a dramatic rearrangement of actin, which is also favored by the actin stabilization and nucleation activities provided by SipA and SipC, two other SPI-1 effectors. Once inside the host cell, Salmonella induces the recovery of normal cytoskeleton dynamics via SptP, a SPI-1 effector with GAP activity that returns Cdc42 and Rac1 to the non-activated state [54]. The interplay between the bacterial effectors acting as GEFs and GAPs (SopE/SptP) is modulated by the proteasome [34]. Both SopE and SptP are delivered into the cytosol in equal amounts early during invasion but SopE is rapidly degraded by the ubiquitin-proteasome system. SptP shows higher resistance to such degradation and down-regulates Cdc42 and Rac1 once SopE is degraded. Lastly, disruption of intermediate filaments, such as cytokeratins, mediated by SipC also seems to be involved in Salmonella entry [7].
Shigella uses a trigger mechanism that shares some features with that of Salmonella, but which also differs. For example, the host surface molecules β1-integrins and CD44 (hyaluronic acid receptor) are needed for Shigella entry. IpaB and IpaC, two TTSS-secreted proteins required for pore formation, interact with these two eukaryotic surface molecules. The interaction is thought to promote pore formation for delivery of other TTSS effectors. Some authors have also suggested that the IpaBC-integrin/CD44 interaction triggers intracellular signaling cascades [52]. Shigella, similar to Salmonella, targets GTPases of the Rho subfamily. Functional studies have established that Cdc42, Rac1 and Rho are all required for bacterial uptake. This observation is consistent with the formation of filopodia, lamelopodia and stress fibers, triggered by activated Cdc42, Rac and Rho, respectively, in the membrane ruffling area containing the invading Shigella. This area has also been shown to be depleted of PtIns(4,5)P2. IpaC is required for activation of these three GTPases, whereas another TTSS secreted protein, IpaA, down-regulates the extension of the membrane ruffling by binding vinculin. The IpaA-vinculin complex then acts as an actin-depolymerization device, leading to formation of focal-adhesion-like structures and the disappearance of filopodia and stress fibers [52]. Other key processes include the recruitment of ezrin and Src-tyrosine kinase to the area of bacterial contact, which occurs once Rho is activated. Src is further involved in down-regulating Rho activity. Another piece of complexity in this bacterial-host cross-talk has been recently added with the finding that the effector protein VirA promotes microtubule destabilization by binding to tubulin [53]. This alteration is apparently essential for proper membrane ruffling activity and Shigella invasion.
Intracellular lifestyles
Once inside the host cell, intracellular bacteria are contained inside a membrane-bound vacuolar compartment, the phagosome, which is altered by the pathogen for its own benefit. In most cases, the pathogen-containing phagosomes do not follow the route of phagosomes containing inert particles (latex beads, killed microorganisms) or non-pathogenic bacteria [16]. The phagosome maturation process is regulated by signaling routes triggered by Toll-like receptors (TLR) [3], which in turn modulate the activity of host proteins involved in vesicular trafficking. Pathogens may target these routes to ensure that their phagosomes do not undergo sequential fusion with early endosomes, late endosomes and lysosomes, a pathway representing the normal maturation route followed by phagosomes containing inert particles[16,38]. Other intracellular bacterial pathogens have developed an alternate strategy consisting of rapid lysis of the phagosomal membrane, which enables them to reach the host cytosol [38]. Both strategies are outlined in Fig. 2.

Life in the cytosol: vacuolar lysis. A way of avoiding exposure to the degradative compartments of the endocytic route is to escape from the phagosome and colonize the nutrient-rich host cytoplasm. This strategy is followed by Listeria, Shigella, and Rickettsia. These three intracellular pathogens are rapidly propelled within the cytoplasm by triggering the formation of an actin tail in one of the bacterial poles. The membrane-damaging enzymes secreted by the pathogen promote lysis of the phagosomal membrane. IpaB, which also participates in Shigella entry (see above), is essential for phagosome lysis. Several Listeria enzymes act synergistically to lyse the phagosomal membrane, including the pore-forming toxin listeriolysin-O (LLO), a phosphatidyl-inositol-specific phospholipase (PI-PLC) and a broad-spectrum phosphatidyl-choline phospholipase (PC-PLC) [43]. LLO has an optimal activity at the acidic pH of 5.9, which suggests that the Listeria-containing phagosome must undergo some transient maturation (acidification) for the bacterial enzymes to become fully active. A host cell molecule targeted by Listeria is Rab5, a GTPase that controls early endosome trafficking. Macrophage phagosomes containing LLO-negative Listeria mutants are enriched in Rab5 and there is an apparent correlation between the amount of Rab-5 associated with the phagosome and the bactericidal activity of the macrophage [44]. Whether wild-type Listeria excludes Rab5 from the phagosomal membrane to prevent entry into the normal maturation route of the phagosome is at present unknown. Once free in the cytosol, Listeria grows rapidly. An inducible bacteria-encoded hexose-phosphate transporter (Hpt), which shows homology to mammalian transporters, is essential for intracellular growth [9]. This is a clear example of mimicry of a host molecule, which enables the pathogen to hijack a mechanism of nutrient (hexose-phosphate) acquisition. Much less is known about the factors that allow Rickettsia to escape from the phagosomal membrane. A recent study reported that Francisella tularensis is able to gain access to the macrophage cytosol [22]. As with Rickettsia, the F. tularensis factors involved in the process remain to be identified.
As mentioned above, recruitment of actin at one pole of the bacterium has been exploited by some of the pathogens that persist in the cytosol. The formation of this unique actin tail allows the pathogen to infect nearby host cells while maintaining its intracellular location. The IcsA and ActA proteins from Shigella and Listeria, respectively, promote actin polymerization. IcsA binds and activates the neuronal Wiskott-Aldrich syndrome protein (N-WASP), which further facilitates binding of the seven-protein eukaryotic Arp2/3 complex, involved in de novo actin nucleation [27]. ActA mimics N-WASP, directly recruiting the Arp2/3 complex, actin monomers and vasodilator-stimulated phosphoprotein (VASP) [21,27]. The Rickettsia coronii RickA protein has been recently shown to promote actin-tail formation by recruiting and activating the Arp2/3 complex [24].
Intravacuolar lifestyle. Many intracellular bacteria that live inside vacuoles avoid fusion to lysosomes [49]. The only exception known is the obligate intracellular pathogen Coxiella burnetii, which inhabits an acidified lysosomal-like compartment [38]. Fusion with the lysosome is avoided by altering the composition of the phagosomal membrane, which becomes virtually non-fusogenic, or segregated, from the endocytic route. Alternatively, the phagosome can undergo transient fusion events with upper compartments of the endocytic route, thereby becoming "arrested" for further maturation steps.
Segregation from the endocytic route. Chlamydia, Legionella, and Brucella inhabit phagosomes that are segregated from the endocytic route. The most extreme case is that of Chlamydia, which resides in a membrane-bound compartment, termed an inclusion, that traffics to the perinuclear region in close proximity to the Golgi apparatus. The inclusion does not intersect with any of the endocytic compartments during this process. The host cell motor protein dynein is hijacked for movement of the inclusion along microtubules [26]. Once in proximity to the Golgi, exocytic vesicles containing sphingomyelin fuse with the Chlamydia inclusion. These observations suggest that Chlamydia programs the unique inclusion compartment upon bacterial entry, at an early post-infection stage. A recent study has shown that Rab1, Rab4 and Rab11, small GTPases involved in receptor recycling (Rab4 and Rab11) and endoplasmic reticulum (ER)-Golgi trafficking (Rab1), are present in the Chlamydia inclusion [47]. These data favor a selective fusion of the inclusion with the exocytic machinery of the host cell. Accordingly, the Chlamydia inclusion does not acquire other Rab proteins involved in endocytic traffic, such as Rab5, Rab7 and Rab9 [47]. Chlamydia protein candidates for these alterations include IncA, IncB, and IncC, which are delivered with the inclusion by a specialized type III secretion system.
Similar to Chlamydia, the Legionella-containing phagosome segregates from the endocytic route. However, in this case the phagosomal compartment is sequentially surrounded by smooth vesicles, mitochondria and the ER [49]. Whether the phagosomal membrane is lost and the pathogen then proliferates in a rough-ER-like compartment is still unknown. During its biogenesis, the Legionella phagosome does not acquire late endosome/lysosome markers such as Rab7 and lysosomal-membrane glycoproteins (Lgps). A Legionella membrane protein, DotA, is essential for segregation of the phagosome from the endocytic route, but how DotA achieves this function is also unknown. Another Legionella protein, RalF, which has a motif conserved in nucleotide exchange factors, recruits the host protein ARF-1 to the phagosomal membrane [39]. ARF-1 has been implicated in the traffic from ER to Golgi. Retention of ARF-1 may then redirect trafficking and contribute to the formation of the unique Legionella phagosome.
Upon infection of epithelial cells, Brucella are initially routed to early endosomal compartments positive for the markers Rab5 and EEA1. However, the Brucella phagosome does not follow the late endosomal/lysosomal route from this early stage. Instead, it traffics to the ER following the autophagocytic route. Host markers that sequentially appear in the phagosome include LAMP1 (late endosome marker), Sec61b (autophagosome marker) and the ER markers sec1b, calnexin and ribophorin [23]. Therefore, like Legionella, Brucella apparently builds an ER-derived compartment permissive for pathogen proliferation. A similar type of compartment has also been described in macrophages. In this infection model, the VirB-dependent secretory system of Brucella is essential for segregating the phagosome from the endocytic route [8].
Arrest of phagosome maturation. Salmonella and Mycobacterium arrest the maturation of the phagosome at specific stages of the phago-lysosomal route [5,12]. In epithelial cells, the Salmonella-containing vacuole (SCV) transiently acquires the early endosome markers Rab5, EEA1 and transferrin-receptor. This stage is followed by acquisition of Lgps, but not lysosomal enzymes, and the subsequent formation of an intricate network of Lgp-containing filamentous structures. Acquisition of Lgps by SCV is dependent on a functional Rab7 protein, which is located in vacuoles surrounding the SCV [31]. Therefore, the SCV seems to rapidly transient through the early endosome and to further specialize in a compartment that acquires late endosomal markers but avoids fusion with lysosomes. Replacement of Rab5 by Rab7 seems to be critical for this process since overexpression of Rab5 impairs Lgps acquisition by the SCV (reviewed in [31]). Experiments using fluid endocytic tracers corroborate this scheme of maturation arrest.
The TTSS encoded in the Salmonella-pathogenicity island-2 (SPI-2) is critical for SCV remodeling. The absence of a functional SPI-2 TTSS causes defects in intracellular proliferation and survival that are more evident in the macrophage infection model [30]. The SPI-2 effector SifA (encoded by a gene mapping outside SPI-2) is required for maintaining the integrity of the SCV membrane. Other SPI-2 effectors, such as SseJ (also encoded outside SPI2), display similarities to eukaryotic lipases. In the absence of SifA, SseJ activity might be deregulated, resulting in lysis of the SCV membrane. Another SPI-2 protein, SpiC, has been shown by in vitro assays to inhibit SCV-lysosome fusion, and two eukaryotic proteins involved in vesicular trafficking, Hook3 and TassC, have been reported to be SpiC targets [36,50]. The classical model of the Salmonella SCV intracellular trafficking route has been slightly modified. Thus, both fusion of the SCV with compartments containing lysosomal enzymes (as cathepsin-D) and accessibility of the compartment to fluid-endocytic markers seem to occur at late infection times [5,49]. Cholesterol also accumulates in the SCV, a process dependent on a functional TTSS encoded by SPI-1 [31]. SseG, a SPI-2 secreted protein, has also been shown to target the trans-Golgi network (TGN) [48], an essential step for the onset of intracellular bacterial proliferation. Taken together, these observations indicate that the SCV is a highly dynamic compartment that undergoes transient interactions with both the endocytic and exocytic routes.
The Mycobacterium phagosome is characterized by the retention of Rab5 and exclusion of the late-endosomal GTPase Rab7 [12]. The host proton-vacuolar ATPase is also excluded, which is consistent with the non-acidic pH estimated for this phagosomal compartment [46]. Interestingly, EEA1, a protein that binds to Rab5, is not observed in the Mycobacterium phagosome. The lipoarabinomannan released from the Mycobacterium envelope might be responsible for this alteration [46]. Another protein with a role in biogenesis of the Mycobacterium phagosome is tryptophane-aspartate containing coat (TACO, also known as coronin). Phagosomes containing viable mycobacteria are decorated with this host protein, which otherwise is not observed in either phagosomes containing killed bacteria or phagosomes of activated macrophages. Therefore, TACO is apparently hijacked by Mycobacterium to arrest phagosome maturation, probably by impairing recruitment of vacuolar-ATPases and other host proteins that direct the phagosome to the lysosome fusion stage.
Hijacking of host lipid microdomains ("lipid rafts")
Certain intracellular bacterial pathogens manipulate host lipid metabolism not only to promote bacterial uptake but also to remodel the vacuolar compartment where they are contained [4]. Targeting of membrane microdomains enriched in sphingolipids and cholesterol, termed "lipid rafts", has been the focus of much interest. A protein present selectively in lipid rafts, flotillin-1, has been detected in phagosomes containing inert particles and is thought to be involved in the acquisition of vacuolar-pump ATPases. Intracellular eukaryotic parasites such as Leishmania inhibit the presence of lipid rafts in the parasitophorous vacuole [13]. Proteomic studies have revealed that lipid rafts from phagosomes contain components of machineries involved in vesicular trafficking and maturation of the compartment: Rab1, Rab7, SNAP-23, the ER marker calnexin, the late endosomal/lysosomal marker LIMP-II, the cytoskeletal proteins actin, α-actinin and vimentin, and regulatory proteins that modulate cytoskeleton dynamics as various Rac isoforms [37]. Interestingly, intracellular Salmonella secrete effector proteins to detergent-resistant microdomains in internal membranes [32], a process that may contribute to alteration of phagosome maturation.
Phagosome proteomics
New methodologies that allow efficient identification of proteins from complex mixtures are providing new insights as to how phagosomes are modulated by pathogens. Using a two-dimensional and MALDI-TOF/mass spectrometry approach, Desjardins et al. determined the protein profile of latex-bead-containing phagosomes [6]. More than 140 proteins were identified along different time points of the latex-bead-phagosome maturation process. Strikingly, ER-resident proteins such as calnexin were identified, suggesting that the phagosomal compartment undergoes fusion with the ER. It was later shown that the ER fuses transiently with the plasma membrane in the very initial phase of phagocytosis of latex beads [19]. This ER-nascent phagosome fusion event also occurs during phagocytosis of bacteria, a process that has been shown to support antigen cross-presentation [14].
Phagosomes containing beads coated with either InlA or InlB from Listeria have also been characterized by a proteomic approach [42]. A protein associated specifically with the InlB-phagosome is MSF, a GTPase from the septin family related to the cytoskeleton. MSF is recruited at the site of entry of InlB-coated beads and forms filaments that co-localize with actin. This study represented an example of how proteomics can identify novel host molecules targeted by the pathogen. Another proteomic study reported differences in the composition of the Francisella-tularensis-containing phagosome depending on the source of macrophages (from mice resistant or sensitive to infection). The differences were found in a putative bacterial protein of 23 kDa, the 60-kDa chaperonin GroEL and a host protein highly homologous to NADH-ubiquinone oxidoreductase [33]. The aforementioned TACO/Coronin1 protein present in the Mycobacterium phagosome was also identified in a proteomic analysis [18].
Genome expression in infections caused by intracellular bacterial pathogens
DNA microarray technology has increased our understanding of host-pathogen interactions [15]. Currently, the complete sequences of 171 microbial (bacteria and archaea) genomes have been obtained (http://www.ncbi.nlm.nih.gov:80/ genomes/MICROBES/Complete.html). Their relatively small sizes make the construction of whole-genome microarrays an affordable task. When analyzing the expression of genomes from bacterial pathogens, a series of variables need to be considered. Thus, gene expression is monitored either under laboratory conditions mimicking the environment most likely encountered by the pathogen in the host or during the infection of susceptible animal hosts or eukaryotic cell types. The potential of these new approaches is exemplified by the recent finding that Chlamydia trachomatis expresses very early in the intracellular stage a gene, named CT147, whose product has homology to human EEA1, involved in early endosomal trafficking [2]. CT147 was proposed as a factor involved in the unique biogenesis process of the Chlamydia inclusion.
Host genome expression has also been examined, especially in comparative studies using different types of pathogens. The effects of bacterial infection on the host eukaryotic cell have been deciphered using pathogens such as Yersinia, Neisseria, Helicobacter pylori, Salmonella, Mycobacterium, and Listeria. Gene expression of infected host cells is often compared with that of uninfected cells or cells infected with isogenic bacterial mutants. A feature observed in many of these studies is that host cells respond to the infection by up-regulating genes encoding surface receptors, cytokines, chemokines, adhesion molecules and transcriptional regulators. In some cases, induction of pro- or anti-apoptotic pathways has also been reported. The reader is referred to Table 2 for a complete list of infection models with bacterial pathogens in which genome expression (host or pathogen) has been analyzed.

Concluding remarks
In this review, the most relevant cases of hijacking of host cellular functions by intracellular bacterial pathogens has been summarized. A common strategy used by these pathogens is modulation of the activity of key host molecules, including surface receptors and regulatory/cytoskeletal proteins. In some instances, the pathogen uses weapons that mimic eukaryotic functions. These weapons manipulate, in a negative or positive fashion, a host regulatory cycle (e.g. GEFs and GAPs of small GTP-binding proteins of the Rho and Rab subfamilies), recruit host protective molecules (e.g. phagosomal-decorating protein TACO), or exclude host factors involved in phagosome-lysosome maturation (Rab proteins from the phagosomal membrane). Despite the great amount of information as to how intracellular bacteria manipulate the host, there are still many impressive phenomena that have only been characterized at a morphological level. These include the unique ER-derived phagosomal compartment built by Legionella and Brucella in order to proliferate inside the infected cell. Chlamydia offers another example of a unique process, biogenesis of the inclusion compartment, which is not understood at the molecular level. The new technologies of genome-wide expression analysis are opening new avenues of research, and recent studies have provided new examples of pathogen proteins used for hijacking specific host functions. Furthermore, these technologies also have the advantage of examining the response to infection of the other partner, the host cell. Information on the host response is equally relevant for deciphering the complex host-pathogen cross-talk and the emergence of these highly specialized group of bacteria.
Acknowledgements. Research in our laboratory on the Salmonella-eukaryotic cell interaction is supported by grants from the Spanish Ministry of Science and Technology (BIO2001-0232-C02 and BIO2001-5243-E) and Comunidad de Madrid (08.2/0019/2003). A. Alonso was supported by a post-doctoral fellowship from the Comunidad de Madrid.
References
1. Alrutz MA, Srivastava A, Wong KW, D'Souza-Schorey C, Tang M, Ch'Ng LE, Snapper SB, Isberg RR (2001) Efficient uptake of Yersinia pseudotuberculosis via integrin receptors involves a Rac1-Arp 2/3 pathway that bypasses N-WASP function. Mol Microbiol 42:689-703 [ Links ]
2. Belland RJ, Zhong G, Crane DD, Hogan D, Sturdevant D, Sharma J, Beatty WL, Caldwell HD (2003) Genomic transcriptional profiling of the developmental cycle of Chlamydia trachomatis. Proc Natl Acad Sci USA 100:8478-8483 [ Links ]
3. Blander JM, Medzhitov R (2004) Regulation of phagosome maturation by signals from Toll-like receptors. Science 304:1014-1018 [ Links ]
4. Brumell JH, Grinstein S (2003) Role of lipid-mediated signal transduction in bacterial internalization. Cell Microbiol 5:287-297 [ Links ]
5. Brumell JH, Grinstein S (2004) Salmonella redirects phagosomal maturation. Curr Opin Microbiol 7:78-84 [ Links ]
6. Brunet S, Thibault P, Gagnon E, Kearney P, Bergeron JJ, Desjardins M (2003) Organelle proteomics: looking at less to see more. Trends Cell Biol 13:629-638 [ Links ]
7. Carlson SA, Omary MB, Jones BD (2002) Identification of cytokeratins as accessory mediators of Salmonella entry into eukaryotic cells. Life Sci 70:1415-1426 [ Links ]
8. Celli J, de Chastellier C, Franchini DM, Pizarro-Cerda J, Moreno E, Gorvel JP (2003) Brucella evades macrophage killing via VirB-dependent sustained interactions with the endoplasmic reticulum. J Exp Med 198:545-556 [ Links ]
9. Chico-Calero I, Suárez M, González-Zorn B, Scortti M, Slaghuis J, Goebel W, The European Listeria Genome Consortium, Vazquez-Boland JA (2002) Hpt, a bacterial homolog of the microsomal glucose-6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc Natl Acad Sci USA 99:431-436 [ Links ]
10. Cornelis GR (2002) The Yersinia Ysc-Yop "type III" weaponry. Nat Rev Mol Cell Biol 3:742-754 [ Links ]
11. Cossart P, Pizarro-Cerda J, Lecuit M (2003) Invasion of mammalian cells by Listeria monocytogenes: functional mimicry to subvert cellular functions. Trends Cell Biol 13:23-31 [ Links ]
12. Deretic V, Fratti RA (1999) Mycobacterium tuberculosis phagosome. Mol Microbiol 31:1603-1609 [ Links ]
13. Dermine JF, Duclos S, Garin J, St-Louis F, Rea S, Parton RG, Desjardins M (2001) Flotillin-1-enriched lipid raft domains accumulate on maturing phagosomes. J Biol Chem 276:18507-18512 [ Links ]
14. Desjardins M (2003) ER-mediated phagocytosis: a new membrane for new functions. Nat Rev Immunol 3:280-291 [ Links ]
15. Diehn M, Relman DA (2001) Comparing functional genomic datasets: lessons from DNA microarray analyses of host-pathogen interactions. Curr Opin Microbiol 4:95-101 [ Links ]
16. Duclos S, Desjardins M (2000) Subversion of a young phagosome: the survival strategies of intracellular pathogens. Cell Microbiol 2:365-377 [ Links ]
17. Eitel J, Dersch P (2002) The YadA protein of Yersinia pseudotuberculosis mediates high-efficiency uptake into human cells under environmental conditions in which invasin is repressed. Infect Immun 70:4880-4891 [ Links ]
18. Fratti RA, Vergne I, Chua J, Skidmore J, Deretic V (2000) Regulators of membrane trafficking and Mycobacterium tuberculosis phagosome maturation block. Electrophoresis 21:3378-3385 [ Links ]
19. Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele-Mortimer O, Paiement J, Bergeron JJ, Desjardins M (2002) Endoplasmic reticulum-mediated phagocytosis is a mechanism of entry into macrophages. Cell 110:119-131 [ Links ]
20. Galan JE (2001) Salmonella interactions with host cells: type III secretion at work. Annu Rev Cell Dev Biol 17:53-86 [ Links ]
21. Goldberg MB (2001) Actin-based motility of intracellular microbial pathogens. Microbiol Mol Biol Rev 65:595-626 [ Links ]
22. Golovliov I, Baranov V, Krocova Z, Kovarova H, Sjostedt A (2003) An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect Immun 71:5940-5950 [ Links ]
23. Gorvel JP, Moreno E (2002) Brucella intracellular life: from invasion to intracellular replication. Vet Microbiol 90:281-297 [ Links ]
24. Gouin E, Egile C, Dehoux P, Villiers V, Adams J, Gertler F, Li R, Cossart P (2004) The RickA protein of Rickettsia conorii activates the Arp2/3 complex. Nature 427:457-446 [ Links ]
25. Grassl GA, Kracht M, Wiedemann A, Hoffmann E, Aepfelbacher M, von Eichel-Streiber C, Bohn E, Autenrieth IB (2003) Activation of NF-κB and IL-8 by Yersinia enterocolitica invasin protein is conferred by engagement of Rac1 and MAP kinase cascades. Cell Microbiol 5:957-971 [ Links ]
26. Grieshaber SS, Grieshaber NA, Hackstadt T (2003) Chlamydia trachomatis uses host cell dynein to traffic to the microtubule-organizing center in a p50 dynamitin-independent process. J Cell Sci 116:3793-3802 [ Links ]
27. Gruenheid S, Finlay BB (2003) Microbial pathogenesis and cytoskeletal function. Nature 422:775-781 [ Links ]
28. Hauck CR, Meyer TF (2003) "Small" talk: Opa proteins as mediators of Neisseria-host-cell communication. Curr Opin Microbiol 6:43-49 [ Links ]
29. Hernández LD, Hueffer K, Wenk MR, Galán JE (2004) Salmonella modulates vesicular traffic by altering phosphoinositide metabolism. Science 304:1805-1807 [ Links ]
30. Holden DW (2002) Trafficking of the Salmonella vacuole in macrophages. Traffic 3:161-169 [ Links ]
31. Knodler LA, Steele-Mortimer O (2003) Taking possession: biogenesis of the Salmonella-containing vacuole. Traffic 4:587-599 [ Links ]
32. Knodler LA, Vallance BA, Hensel M, Jackel D, Finlay BB, Steele-Mortimer O (2003) Salmonella type III effectors PipB and PipB2 are targeted to detergent-resistant microdomains on internal host cell membranes. Mol Microbiol 49:685-704 [ Links ]
33. Kovarova H, Halada P, Man P, Golovliov I, Krocova Z, Spacek J, Porkertova S, Necasova R (2002) Proteome study of Francisella tularensis live vaccine strain-containing phagosome in Bcg/Nramp1 congenic macrophages: resistant allele contributes to permissive environment and susceptibility to infection. Proteomics 2:85-93 [ Links ]
34. Kubori T, Galan JE (2003) Temporal regulation of Salmonella virulence effector function by proteasome-dependent protein degradation. Cell 115:333-342 [ Links ]
35. Kwok T, Backert S, Schwarz H, Berger J, Meyer TF (2002) Specific entry of Helicobacter pylori into cultured gastric epithelial cells via a zipper-like mechanism. Infect Immun 70:2108-2120 [ Links ]
36. Lee AH, Zareei MP, Daefler S (2002) Identification of a NIP-SNAP homologue as host cell target for Salmonella virulence protein SpiC. Cell Microbiol 4:739-750 [ Links ]
37. Li N, Mak A, Richards DP, Naber C, Keller BO, Li L, Shaw AR (2003) Monocyte lipid rafts contain proteins implicated in vesicular trafficking and phagosome formation. Proteomics 3:536-548 [ Links ]
38. Meresse S, Steele-Mortimer O, Moreno E, Desjardins M, Finlay B, Gorvel JP (1999) Controlling the maturation of pathogen-containing vacuoles: a matter of life and death. Nat Cell Biol 1:E183-188 [ Links ]
39. Nagai H, Kagan JC, Zhu X, Kahn RA, Roy CR (2002) A bacterial guanine nucleotide exchange factor activates ARF on Legionella phagosomes. Science 295:679-682 [ Links ]
40. Niebuhr K, Giuriato S, Pedron T, Philpott DJ, Gaits F, Sable J, Sheetz MP, Parsot C, Sansonetti PJ, Payrastre B (2002) Conversion of PtdIns(4,5)P2 into PtdIns(5)P by the S. flexneri effector IpgD reorganizes host cell morphology. EMBO J 21:5069-5078 [ Links ]
41. Nougayrede JP, Fernandes PJ, Donnenberg MS (2003) Adhesion of enteropathogenic Escherichia coli to host cells. Cell Microbiol 5:359-372 [ Links ]
42. Pizarro-Cerda J, Jonquieres R, Gouin E, Vandekerckhove J, Garin J, Cossart P (2002) Distinct protein patterns associated with Listeria monocytogenes InlA- or InlB-phagosomes. Cell Microbiol 4:101-115 [ Links ]
43. Portnoy DA, Auerbuch V, Glomski IJ (2002) The cell biology of Listeria monocytogenes infection: the intersection of bacterial pathogenesis and cell-mediated immunity. J Cell Biol 158:409-414 [ Links ]
44. Prada-Delgado A, Carrasco-Marin E, Bokoch GM, Alvarez-Dominguez C (2001) Interferon-gamma listericidal action is mediated by novel Rab5a functions at the phagosomal environment. J Biol Chem 276:19059-19065 [ Links ]
45. Rosenberger CM, Finlay BB (2003) Phagocyte sabotage: disruption of macrophage signalling by bacterial pathogens. Nat Rev Mol Cell Biol 4:385-396 [ Links ]
46. Russell DG (2003) Phagosomes, fatty acids and tuberculosis. Nat Cell Biol 5:776-778 [ Links ]
47. Rzomp KA, Scholtes LD, Briggs BJ, Whittaker GR, Scidmore MA (2003) Rab GTPases are recruited to chlamydial inclusions in both a species-dependent and species-independent manner. Infect Immun 71:5855-5870 [ Links ]
48. Salcedo SP, Holden DW (2003) SseG, a virulence protein that targets Salmonella to the Golgi network. EMBO J 22:5003-5014 [ Links ]
49. Scott CC, Botelho RJ, Grinstein S (2003) Phagosome maturation: a few bugs in the system. J Membr Biol 193:137-152 [ Links ]
50. Shotland Y, Kramer H, Groisman EA (2003) The Salmonella SpiC protein targets the mammalian Hook3 protein function to alter cellular trafficking. Mol Microbiol 49:1565-1576 [ Links ]
51. Terebiznik MR, Vieira OV, Marcus SL, Slade A, Yip CM, Trimble WS, Meyer T, Finlay BB, Grinstein S (2002) Elimination of host cell PtdIns(4,5)P(2) by bacterial SigD promotes membrane fission during invasion by Salmonella. Nat Cell Biol 4:766-773 [ Links ]
52. Tran Van Nhieu G, Bourdet-Sicard R, Dumenil G, Blocker A, Sansonetti PJ (2000) Bacterial signals and cell responses during Shigella entry into epithelial cells. Cell Microbiol 2:187-193. [ Links ]
53. Yoshida S, Sasakawa C (2003) Exploiting host microtubule dynamics: a new aspect of bacterial invasion. Trends Microbiol 11:139-143 [ Links ]
54. Zhou D, Galan J (2001) Salmonella entry into host cells: the work in concert of type III secreted effector proteins. Microbes Infect 3:1293-1298 [ Links ]

