










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkMedicina Oral, Patología Oral y Cirugía Bucal (Internet)
versión On-line ISSN 1698-6946
Med. oral patol. oral cir.bucal (Internet) vol.11 no.1 ene./feb. 2006
Enfermedad de Adamantiades-Behçet: Un proceso enigmático
con manifestaciones orales
Asier Eguia 1, Mariana Villarroel 2, Rafael Martínez-Conde 1, María Ángeles Echebarría 1,
José Manuel Aguirre 1
(1) Medicina Bucal. Unidad de Patología Oral y Maxilofacial. Departamento de Estomatología.
Universidad del País Vaso / EHU
(2) Instituto de Investigaciones Odontológicas. Universidad Central de Venezuela
Dirección para correspondencia
RESUMEN
La enfermedad de Adamantiades-Behçet es una vasculitis multisistémica crónica, potencialmente capaz de afectar a cualquier órgano o sistema del cuerpo humano y en la que la aparición repetida de úlceras orales es una de sus principales expresiones clínicas. La EAB es una patología de carácter universal, con una prevalencia variable en función de la población estudiada y que muestra una curiosa distribución geográfica. A pesar de ser un proceso conocido desde la antigüedad, su etiopatogenia en la que probablemente se hayan implicados factores genéticos, microbiológicos e inmunológicos, continua siendo enigmática. Su amplio espectro de manifestaciones clínicas orales, genitales, cutáneas, oculares, neurológicas, vasculares y gastrointestinales y su impredecible evolución con periodos de exacerbación y de remisión son dos de los aspectos más representativos de esta patología. El complejo tratamiento de la EAB requiere una estrecha cooperación multidisciplinar dado su carácter multisistémico. Gracias a ello y al desarrollo de nuevos agentes terapéuticos su pronóstico ha mejorado sensiblemente con respecto a décadas pasadas. En esta revisión analizamos los principales aspectos etiopatogénicos, clínicos y terapéuticos de esta enfermedad.
Palabras clave: Enfermedad de Adamantiades-Behçet, enfermedad de Behçet, úlceras orales, aftas, vasculitis.
ABSTRACT
Adamantiades-Behçet disease (ABD) is a chronic multisystemic vasculitis that is able to affect any human organ or system.
Recurrent oral ulcers are a very important clinical sign. ABD is a worldwide pathology, which prevalence varies according to the population and geographic location. Although ABD has been known for ages, its aetiology remains an enigma. Genetic, immunological and microbiological factors have been associated. A wide spectrum of clinical manifestations (oral, genital, cutaneous, ocular, neurological, vascular and gastrointestinal) and an unpredictable evolution with repeated periods of exacerbation and remission are the most representative aspects of this pathology. The complex treatment of ABD requires a deep multidisciplinary cooperation; therefore, there is an extensive development of new therapeutic agents that have improved the prognosis of ABD. In this review were analysed the main etiological, clinical and therapeutic aspects of the disease.
Key words: Adamantiades-Behçet disease, Behçet disease, oral ulcers, aphthous stomatitis, vasculitis.
Introducción
La enfermedad de Adamantiades-Behçet (EAB) es un proceso multisistémico, de etiología desconocida, cuyo mecanismo patogénico puede ser una disregulación inmunitaria en la que la vasculitis es su principal expresión (1,2). Las ulceras orales recurrentes son la manifestación clínica más frecuente de esta patología. No obstante, la vasculitis que la caracteriza es potencialmente capaz de afectar a prácticamente todos los órganos y sistemas del organismo, por consiguiente los rasgos de esta patología pueden ser múltiples, variados y diferentes en cada paciente (1-3).
Aunque reconocida por primera vez por Hipócrates en el siglo V a.C., no existen referencias sobre esta enfermedad hasta 1931, cuando el oftalmólogo griego Benedict Adamantiades realizó la primera descripción clínica (1,4). Posteriormente y de forma independiente, en 1937, fue el dermatólogo turco Hulusi Behçet quien realizó una descripción completa de la enfermedad y definió su clínica con la clásica triada "ulceración oral, genital y lesiones oculares" (1,4). Diversas investigaciones posteriores permitieron asociar a su espectro clínico las lesiones cutáneas, articulares, digestivas, vasculares y neurológicas.
En memoria a su contribución, algunos autores (4) consideran más correcto nombrar a este proceso "enfermedad de Adamantiades-Behçet", que sólo "enfermedad de Behçet". También es controvertido el si se debe denominar "enfermedad" o "síndrome", ya que para unos posee la suficiente entidad clínica como para denominarlo "enfermedad", aunque para otros, el desconocimiento de su etiología y su gran variabilidad clínica aconsejan denominarlo "síndrome" (1,2).
Epidemiología
La EAB es una patología de carácter universal, aunque muestra una curiosa distribución geográfica. Su prevalencia es elevada en el sudeste asiático (2-20/100.000), los países mediterráneos y en los países por los que transcurría la "ruta de la seda" (13-370/100.000). Es especialmente frecuente en Japón (13-30/100.000), Turquía (80-300/100.000) y Corea del Sur, mientras que su prevalencia es baja en los países Europeos no Mediterráneos y en los Estados Unidos (0,1-7,5 casos/100.000) (5-8). Aunque ciertos grupos étnicos han mostrado una mayor predisposición, existe controversia sobre si se encuentra relacionada con el área geográfica o con caracteres raciales (5-8). La EAB afecta por igual a hombres y mujeres, salvo en algunos países de Oriente Medio como Irán, Turquía, Líbano, Irak, Jordania, Israel o Egipto, donde estudios sobre amplios grupos poblacionales han revelado una mayor prevalencia entre los varones (1.5-5:1) (5-8).
El comienzo de los síntomas puede producirse a cualquier edad, no obstante, en la mayor parte de los casos se produce en la tercera década de la vida y habitualmente el espectro clínico completo se desarrolla en los 15 meses posteriores al debut de la enfermedad. Apenas existen casos neonatales publicados y los casos infantiles son poco frecuentes y no muestran grandes diferencias clínicas con respecto a los enfermos adultos (1-3). El comienzo precoz de los síntomas en pacientes varones, se asocia a una mayor severidad clínica (9).
Etiopatogenia
La etiología exacta de la EAB es aún desconocida y difiere del patrón característico de las enfermedades autoinmunes. La existencia de casos familiares documentados, su estrecha relación con determinados genes y su caprichosa distribución geográfica denotan la importancia de un componente genético o ambiental en su etiopatogenia (1,10). No obstante, dado que no sigue un patrón de herencia mendeliana, que la prevalencia de casos familiares es baja (1-18%) y que no se ha encontrado una alteración genética exclusiva, no se puede atribuir su etiología únicamente a factores genéticos (1,10).
Se ha demostrado la estrecha relación de esta patología con el gen del Complejo Mayor de Histocompatibilidad (CMH) HLA-B51, especialmente en pacientes de Japón, Oriente Medio y de la cuenca Mediterránea. Su presencia es más común en varones y también se ha asociado a síntomas oculares (1-3). Otros genes del CMH que han demostrado una estrecha relación con esta condición son el HLA-B27, ligado a la existencia de lesiones articulares y el HLA-B12, asociado a la presencia de manifestaciones mucocutáneas (1-3). En la actualidad se desarrollan múltiples investigaciones en la búsqueda de nuevos factores o marcadores genéticos tratando de conocer su implicación en la EAB como los genes HLA-DRw8, HLA-B15 o MIC-A, o moléculas de adhesión como las variantes del ICAM-1, las cuales podrían estar relacionadas con la vasculitis que se origina en este proceso (11-15).
Desde las primeras descripciones de esta enfermedad se especuló con la posible implicación de agentes infecciosos en su etiología, sobre todo apoyándose en su distribución geográfica. Curiosamente, incluso Behçet en su publicación original, apuntaba un origen vírico de la enfermedad debido al aspecto clínico de las lesiones orales y genitales (16). Diversos estudios (1-3), han tratado de comprobar la existencia de una implicación vírica, especialmente de la familia de los virus herpes, o bacteriana, especialmente de algunos tipos de estreptococos, sin obtener resultados concluyentes. Diferentes autores (1-3,14,15) han identificado ADN de virus herpes simple tipo I en linfocitos y monocitos en sangre y/o anticuerpos séricos en una mayor proporción que en la población general. Sin embargo, la proporción de pacientes con serología positiva, y con material genético viral en las biopsias de las lesiones no fue estadísticamente significativa. Los resultados de ciertos estudios in vitro (1-3,14,15) han hecho especular con la posibilidad de que una cierta hipersensibilidad a determinadas cepas de Streptococcus sanguis podría jugar un papel relevante en la patogénesis de la enfermedad. Incluso el hecho de que la manifestación más común de la enfermedad sean las lesiones orales, podría no ser casual y estar de algún modo en relación con los estreptococos bucales. Por el momento, la falta de evidencias concluyentes impide estimar la relevancia de los agentes infecciosos en la etiopatogenia de la EAB.
Otra pieza en el rompecabezas que constituye la etiopatogenia de la EAB, son las alteraciones inmunológicas que frecuentemente se observan en estos pacientes. Hasta un 50% de pacientes muestran un aumento de inmunocomplejos circulantes durante los periodos de exacerbación de la enfermedad (1-3,10). Algunos estudios (10,14,15) han observado, durante las fases activas de la enfermedad, una disminución del ratio linfocitos CD4/CD8 por aumento de linfocitos CD8, una actividad disminuida de la actividad de células natural killer (NK) o un aumento de los niveles de interleuquina IL-2, IL-10 o factor de necrosis tumoral TNFRγ. También se ha podido apreciar aumento de la concentración de linfocitos T γδ capaces de secretar linfoquinas y desarrollar actividad citolítica (10,14,15). Algunas de las sustancias producidas por los linfocitos T, podrían jugar un papel importante en las aftas orales, como el Factor de Crecimiento Fibroblástico FGF-7 que estimula y acelera la proliferación fibroblástica, y el Factor de Necrosis Tumoral (TNF), que parece jugar un papel fundamental en la génesis de las lesiones orales (10-12).
En ausencia de un consenso generalizado sobre su etiopatogenia, debemos considerar por el momento que la EAB es una condición de etiología multifactorial, que no puede ser clasificada aisladamente como una enfermedad hereditaria, infecciosa, o autoinmune.
Clínica
Son características llamativas de la EAB, su amplio espectro clínico y su impredecible evolución con periodos de exacerbación y de remisión (17). Dentro de sus manifestaciones clínicas se pueden observar lesiones orales, genitales, cutáneas, oculares, neurológicas, vasculares y gastrointestinales.
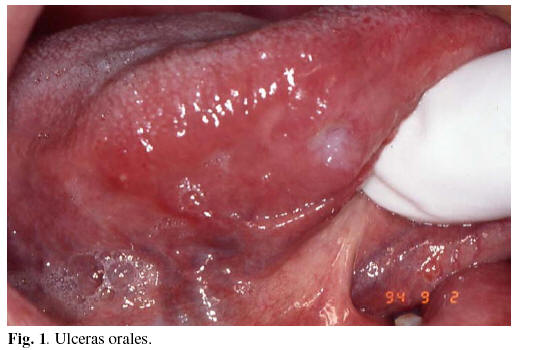
1) Lesiones orales: Están presentes en el 95-100 % de los pacientes (1-3), clínicamente son indistinguibles de las ulceras de la Estomatitis Aftosa Recurrente (EAR). Como en la EAR, las úlceras orales se clasifican en: menores (< 1 cm.), mayores (> 1 cm.) y herpetiformes (1-3 mm) (1,17) (Figura 1). Apenas se ha podido constatar diferencias clínicas entre las lesiones orales en la EAR y en la EAB. No obstante, se indica una cierta tendencia a padecer un mayor número de lesiones y en localizaciones poco habituales (17). Tanto la capacidad de los traumatismos de desencadenar nuevas lesiones como el paradójico efecto protector del tabaco, hipotéticamente basado en la hiperqueratinización que produce, son características comunes contrastadas en ambos procesos (17,18). Las lesiones orales constituyen en muchas ocasiones la primera manifestación de la EAB y en gran parte de los pacientes transcurre entre 1 y 3 años entre la aparición de lesiones orales y el despliegue de su amplio abanico clínico (17,18).
2) Lesiones genitales: Son menos frecuentes que las anteriores (60-80%) y se presentan en forma de ulceras similares a las orales (1-3). Pueden aparecer en cualquier zona del territorio genital, siendo la localización más común el escroto en los varones y los labios mayores y menores en las mujeres. Son dolorosas y pueden provocar problemas para caminar y sentarse y en ocasiones disuria. La evolución de las úlceras genitales puede verse complicada por sobreinfección, cuyo riesgo aumenta cuanto mayor sea el tamaño y la profundidad (1-3,19).
3) Lesiones cutáneas: La EAB provoca la aparición de lesiones cutáneas hasta en el 80% de los pacientes, siendo las lesiones más frecuentes las del tipo eritema nudoso (40-50%) y las lesiones papulo-pustulosas o acneiformes (60-70%) (20). Las lesiones tipo eritema nudoso son más frecuentes en las extremidades inferiores, generalmente no llegan a ulcerarse y pueden dejar un área hiperpigmentada. Histológicamente se corresponden con una vasculitis focal de pequeños vasos (20). Las lesiones pápulo-pustulares o acneiformes pueden aparecer en cualquier localización y son similares morfológicamente a las de la adolescencia. Histológicamente, junto a la vasculitis de pequeños vasos, se aprecia un infiltrado inflamatorio con neutrófilos y necrosis fibrinoide (20).
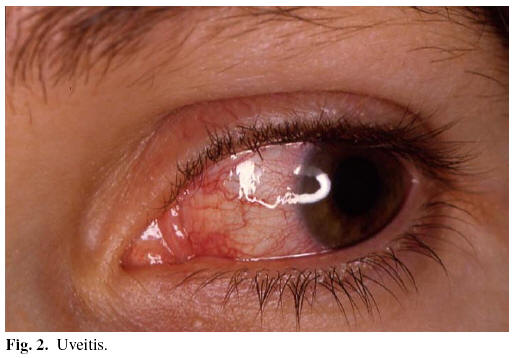
4) Lesiones oculares: Se pueden evidenciar en un 20 a 50% de pacientes con EAB (1-3). Habitualmente no es un signo que aparezca en el inicio de la enfermedad, sino tras 2 o 3 años. Las manifestaciones pueden ser muy variadas; desde las más leves como uveitis o conjuntivitis, cuando son aisladas, hasta vasculitis y atrofia retiniana, glaucoma, cataratas o desprendimiento de retina (17,21) (Figura 2). La severidad de las manifestaciones oculares es variable y generalmente bilateral, observándose pérdida de visión en una cuarta parte de los pacientes e incluso ceguera (21).
5) Lesiones articulares: Hasta en el 45-50% de pacientes con EAB se puede apreciar una artritis no deformante, monoarticular u oligoarticular, que cursa en episodios de varios días o semanas, afectando principalmente a las articulaciones de las extremidades (1-3,17).
6) Lesiones neurológicas: Son apreciables en un 5-20% de pacientes y pueden afectar al cerebro, meninges y médula espinal (22). Clínicamente se manifiestan como alteraciones emocionales, sensoriales, hemiparesias, parestesias, afectación de pares craneales e incluso desordenes psiquiátricos. El líquido cefalorraquideo puede ser normal o mostrar una elevación de la presión, así como un aumento de la concentración de neutrófilos, linfocitos o proteínas (17,22).
7) Lesiones vasculares: En esencia, todas las lesiones asociadas a la EAB son consecuencia directa o indirecta de una vasculitis, que puede producirse en todo tipo de vasos sanguíneos; venosos o arteriales, profundos o superficiales, si bien, es más común en los de pequeño calibre (23). Se ha descrito una mayor tendencia a la trombosis tanto superficial como profunda, especialmente en las extremidades inferiores. Afortunadamente las complicaciones como el embolismo pulmonar son poco comunes. Este hecho ha generado controversia con respecto a la necesidad de emplear una terapia anticoagulante preventiva en la EAB (17,23).En los pacientes con EAB, el riesgo de formación de aneurismas arteriales y de oclusión arterial es también más elevado que en pacientes sanos. Su aparición en arterias de gran calibre, aunque infrecuente, puede acarrear graves consecuencias (23).
8) Lesiones gastrointestinales: La prevalencia de este tipo de lesiones varía ampliamente en dependencia de la población que se estudie, siendo particularmente importante entre los pacientes japoneses (1-3,17). Las principales alteraciones son de tipo ulcerativo, y pueden presentarse en cualquier localización desde el esófago hasta el recto. Los síntomas varían según la localización y gravedad (malestar, disfagia, dolor abdominal, diarrea, etc). Aunque es infrecuente, las lesiones pueden complicarse formándose fístulas, perforaciones, etc (17).
9) Otras lesiones: Existen otras numerosas alteraciones que se han descrito en estos pacientes y cuya incidencia es menor que las anteriormente citadas. Las más frecuentes han sido las complicaciones cardiacas (pericarditis, trombosis coronaria, prolapso valvular), renales (proteinuria, hematuria o glomerulonefritis) y pulmonares y pleurales (17-23).
Diagnóstico
El diagnóstico de la EAB continua siendo difícil en algunas ocasiones; ya que en ausencia de una prueba diagnóstica concluyente que permita confirmar un diagnóstico de presunción, este debe realizarse en base a la clínica (1-3). La gran relevancia que en la antigüedad se otorgaba al "Test de patergia", que mide la reacción que se produce 24 o 48 horas después de una punción intradérmica en el antebrazo (positivo si el nódulo eritematoso o la pústula que se forma es mayor de 2 mm), ha decaído conforme se ha demostrado que hasta en el 50% puede ser negativo (24).
Con el fin de unificar criterios de inclusión y de homogeneizar los grupos de estudio, diversos grupos han establecido diferentes clasificaciones. De ellas, la que quizá goce de una mayor aceptación entre la Comunidad Científica Internacional es la establecida en 1990 por el Grupo de Estudio Internacional para la Enfermedad de Behçet (25). En esta clasificación es condición imprescindible para el diagnóstico la existencia de 3 episodios de aftas orales, ya sean menores, mayores o herpetiformes, dentro de un periodo de 12 meses, y además deben existir en la historia clínica, 2 de entre las 4 siguientes manifestaciones: ulceras genitales de repetición, lesiones oculares, lesiones cutáneas o Test de patergia positivo (25) (Tabla I). La utilidad de estos criterios es mayor en el campo de la investigación que en la práctica diaria, ya que existen pacientes que sin cumplir completamente estos criterios de inclusión, presentan una clínica que coherentemente debería diagnosticarse como EAB. Esto ocurre por ejemplo en algunos casos, en los que no existen o no aparecen con la suficiente periodicidad lesiones orales y sin embargo existen otros múltiples signos y síntomas significativos (24).

Como en cualquier otra patología, o más aún dada la ausencia de pruebas diagnósticas concluyentes, es indispensable realizar una exhaustiva, completa y correcta historia clínica del paciente para poder establecer un correcto diagnóstico diferencial con otros procesos y poder llegar a un diagnóstico final acertado. Dependiendo del tipo de manifestaciones clínicas que se presenten en cada paciente, el diagnóstico diferencial debe realizarse con distintas patologías como: la Estomatitis aftosa recurrente, la Aftosis recurrente oral y genital (Aftosis bipolar de Neumann), el síndrome de Reiter, la sarcoidosis, la esclerosis múltiple, el síndrome de Steven-Johnson, el síndrome PFAPA, el Síndrome MAGIC, la enfermedad de Crohn o la enfermedad celiaca.
Tratamiento
Debido a la afectación que provoca la EAB de diferentes órganos y tejidos, es obvio que el enfoque terapéutico debe ser obligatoriamente multidisciplinario. El correcto tratamiento del paciente con EAB pasa por una estrecha colaboración entre diferentes especialistas, para coordinar un protocolo terapéutico individualizado en cada paciente que combine el tratamiento general del proceso con el tratamiento específico dirigido a cada uno de los órganos o tejidos afectados (26). En el tratamiento general de la EAB y con el fin de atenuar y prevenir los periodos de actividad, se emplean diversos fármacos antinflamatorios, corticoides o inmunomoduladores (26). De entre los nuevos fármacos con los que se experimenta en la actualidad, el interferón alfa 2A y B es el fármaco que muestra resultados más optimistas. Su empleo parece ser eficaz en la reducción de los síntomas y en la prevención de la aparición de brotes de actividad de la enfermedad y su margen de seguridad es aceptable, aunque las dificultades en la estandarización de la dosificación y la escasa experiencia sobre su uso en la EAB son sus mayores inconvenientes (27).
En lo que refiere al tratamiento de las lesiones orales, se emplea básicamente la misma terapéutica que se emplea en los pacientes con EAR, optando en los casos más leves por tratamientos tópicos y reservando los tratamientos sistémicos para los casos más graves (28,29). Dentro de la terapéutica tópica, los esteroides han sido clásicamente la principal arma en el tratamiento de las úlceras orales. La triamcinolona (0,05-0,5%), la fluocinolona (0,05-0,1 %), el clobetasol (0,05-0,1%) y la betametasona (0,1%) permiten reducir considerablemente la sintomatología y duración de las lesiones, aunque no evitan las recurrencias (26-29). La utilidad de otros fármacos utilizados tópicamente como anestésicos, analgésicos, antisépticos o productos cicatrizantes es limitada, por lo que en algunos casos solo se emplean como complemento de otros agentes, bien sólos o dentro de diferentes formulaciones magistrales (28,29).
En caso de lesiones orales severas, se emplean fármacos por vía sistémica, integrando su uso dentro del protocolo terapéutico general y tras sopesar detenidamente la relación riesgo-beneficio que su uso supone. El empleo de fármacos con potentes efectos inmunomoduladores como la talidomida, tacrolimus, azatioprina, azelastina o metotrexate permiten conseguir una reducción del número de lesiones, del dolor, de la duración de los brotes y un aumento del periodo de latencia. Sin embargo el empleo de estos medicamentos puede acompañarse de importantes efectos secundarios como teratogenicidad, trastornos digestivos severos, hematológicos, etc, por lo que durante su uso el paciente debe ser monitorizado y las mujeres en edad fértil deben seguir estrictas medidas anticonceptivas. Su dosificación es individualizada y debe ser estrictamente respetada (26-29). La administración de corticoides sistémicos, como la prednisona en diferentes dosificaciones, consigue una curación rápida de las lesiones y periodos de latencia sensiblemente mayores, aunque su administración debe realizarse bajo estrictas medidas que permitan reducir sus efectos secundarios (28,29). Otros fármacos como la pentoxifilina, fármaco con acciones hemorreológicas e inmunomodulador o antimicóticos como la colchicina permiten reducir e incluso evitar la aparición de nuevos brotes de lesiones durante largos periodos de tiempo incluso tras cesar su administración, aunque no se conoce con exactitud a través de que mecanismo de acción. (26-29). Como en los casos anteriores, su uso debe realizarse bajo una minuciosa dosificación y monitorización de los pacientes.
Pronostico
La evolución de la EAB es crónica y se caracteriza por impredecibles periodos de remisión y de actividad. Generalmente, la frecuencia e intensidad de los periodos de actividad de la enfermedad se reducen con el tiempo, aunque aun no se puede predecir la evolución de estos pacientes. Dependiendo de los órganos y tejidos que se vean afectados, el grado de morbilidad y de secuelas puede variar. Hasta un 20% de pacientes pueden presentar una evolución severa y pueden aparecer complicaciones como: trombosis, perforación intestinal, infarto de miocardio, rotura de aneurismas y otras, que pueden llevar incluso a la muerte del paciente. Afortunadamente son infrecuentes y gracias a la mejora de las terapéuticas preventivas, hoy en día la tasa de mortalidad de los pacientes con EAB es prácticamente similar a la de la población general (30). La morbilidad en cambio, sigue siendo elevada ya que la EAB puede conducir a graves discapacidades como ceguera o artritis severas.
Bibliografía
1. Kontogiannis V, Powell RJ. Behçet´s disease. Postgrad Med J 2000;76: 629-37. [ Links ]
2. Lee LA. Behcet disease. Semin Cutan Med Surg 2001;20:53-7. [ Links ]
3. Barnes CG, Yazici H. Behcet´s Syndrome. Rheumatology 1999;38:1171-6. [ Links ]
4. Dimakakos PB, Tsiligiris B, Kotsis T. The physician B. Adamantiades and his contribution to the disease Adamantiades-Behcet. Int Angiol 1999; 18:176-81. [ Links ]
5. Zouboulis CC. Epidemiology of Adamantiades-Behçet´s disease. Ann Med Int 1999;150:488-98. [ Links ]
6. Jaber L, Milo G, Halpern GJ, Krause I, Weinberger A. Prevalence of Behçet´s disease in an arab community in Israel. Ann Rheum Dis 2002;61:365-6. [ Links ]
7. Verity DH, Marr JE, Ohno S, Wallace GR, Stanford MR. Behçet´s disease, the silk road and HLA-B51: historical and geographical perspectives. Tissue Antigens 1999;54:213-20. [ Links ]
8. Zouboulis C, Kotter I, Djawari D, Kirch W, Kohl P, Ochsendorf FR et al. Epidemiological features of Adamantiades- Behçet´s disease in Germany and in Europe. Yonsei Med J 1997;38:411-22. [ Links ]
9. Yacizi H, Yurdakul S, Tüzün Y. Influence of age and onset and patient´s sex on the prevalence and severity of manifestations of Behçet´s disease. Ann Rheum Dis 1984;43:783-9. [ Links ]
10. Onder M, Gurer MA. The multiple faces of Behçet´s disease and its aetiological factors. J Eur Acad Dermatol Venereol 2000;15:126-36. [ Links ]
11. Lehner T, Welsh KI, Batchelor JR. The relationship of HLA-B and DR phenotypes to Behçet´s syndrome, recurrent oral ulceration and the class of immune complexes. Immunology 1982;47:581-7. [ Links ]
12. Kim EH, Mok JW, Bang D, Lee ES, Lee S, Park K. Intercellular adhesion molecule-1 polymorphisms in Korea patients with Behçet´s disease. J Korean Med Sci 2003;18:415-8. [ Links ]
13. Sun A, Hsieh RP, Chu CT, Wang JT, Liu BY, Chiang CP. Some specific human leukocyte antigen (HLA)-DR/DQ haplotypes are more important than individual HLA-DR and –DQ phenotypes for the development of mucocutaneous type of Behçet´s disease and for disease shift from recurrent aphthous stomatitis to mucocutaneous type of Behçet´s disease. J Oral Pathol Med 2001;30:402-7. [ Links ]
14. Meador R, Ehrlich G, Von Feldt JM. Behçet´s disease: immunopathologic and therapeutic aspects. Curr Rheumatol Rep 2002;4:47-54. [ Links ]
15. Emmi L, Brugnolo F, Marchione T. Pathogenesis and therapy of Behçet´s disease. Ann Ital Med Int 1997;12:20-5. [ Links ]
16.Behçet H. Uber rezidivierende, aphtose, durch ein virus verursachte. Geschwure am Mund, am Auge und an den Genitalen. Dermatol Wochenchir 1937;36:1152-7. [ Links ]
17.Bang D. Clinical spectrum of Behçet´s disease. J Dermatol 2001;28: 610-3. [ Links ]
18. Rizvi SW, McGrath H Jr. The therapeutic effect of cigarette smoking on oral/genital aphtosis and other manifestations of Behçet´s disease. Clin Exp Rheumatol 2001;19:77-8. [ Links ]
19. Cappele O, Nicolas J, Bottet P, Bensadoum H. Urological manifestations of Behçet´s disease. Prog Urol 2003;77:329-31. [ Links ]
20. Lee E, Bang D, Lee S. Dermatologic manifestations of Behçet´s disease. Yonsei Med J 1997;38:380-9. [ Links ]
21. Tugal-Tutkun I, Onal S, Altan-Yaycioglu R, Huseyin Altunbas H, Urgacioglu M. Uveitis in Behçet disease: An analysis of 880 patients. Am J Ophtalmol 2004;138:373-80. [ Links ]
22. Silva A, Altintas A, Saip S. Behcet´s syndrome and the nervous system. Curr Opin Neurol 2004;17:347-57. [ Links ]
23. Koc Y, Güllü I, Akpek G, Akpolat T, Kansu E, Kiraz S et al. Vascular involvement in Behçet´s disease. J Rheumatol 1992;19:402-10. [ Links ]
24. Kyu H, Cheon KS. The clinical significance of a pathergy reaction in patients with Behçet´s disease. J Korean Med Sci 2002;17:371-4. [ Links ]
25. International Study Group for Behçet´s Disease. Criteria for diagnosis of Behçet´s disease. Lancet 1990;335:1078-80. [ Links ]
26. Whallet AJ, Thurairajan G, Hamburger J, Palmer RG, Murray PI. Behçet´s syndrome: a multidisciplinary approach to clinical care. Q J Med 1999;92: 727-40. [ Links ]
27. Kotter I, Gunaydin I, Zierhut M, Stubiger N. The use of interferon alpha in Behcet disease: review of the literature. Semin Arthritis Rheum 2004; 33:320-35. [ Links ]
28. Sakane T, Takeno M. Current therapy in Behçet´s disease. Skin Therapy Lett 2000;5:3-5. [ Links ]
29. Barrons RW. Treatment strategies for recurrent oral aphtous ulcers. Am J Health Syst Pharm 2001;58:41-53. [ Links ]
30. Yacizi H, Basaran G, Hamuryudam V, Hizli N, Yurdakul S, Mat C et al. The ten-year mortality in Behçet´s syndrome. Br J Rheumatol 1996;35:139-41. [ Links ]
![]() Dirección para correspondencia
Dirección para correspondencia
Dr. J.M. Aguirre
Medicina Bucal. Departamento de Estomatología
Facultad de Medicina y Odontología
Universidad del País Vasco EHU
Leioa 48940. Vizcaya.
E-mail: otpagurj@lg.ehu.es
Recibido: 23-05-2004
Aceptado: 30-01-2005

