










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkAvances en Periodoncia e Implantología Oral
versión On-line ISSN 2340-3209versión impresa ISSN 1699-6585
Avances en Periodoncia vol.15 no.3 Madrid dic. 2003
Mecanismos inmunológicos de las enfermedades periodontales y periimplantarias
| BASCONES A * GONZÁLEZ MOLES MA ** | Bascones A, González Moles MA. Mecanismos inmunológicos de las enfermedades periodontales y periimplantarias. Av Periodon Implantol. 2003; 15, 3: 121-138.
|
RESUMEN
Se presenta un trabajo de revisión sobre los mecanismos inmunológicos y el proceso destructivo de la periodontitis. Comenzando con los conceptos básicos sobre la inmunidad natural y adquirida. Se hace un estudio sobre el reconocimiento antigénico mediadores de la inflamación, líneas de defensa del organismo y factores de riesgo implicados en la enfermedad. Continua el trabajo con un estudio sobre el mecanismo apoptótico y una valoración sobre las metaloproteinasas liberadas y responsables de la destrucción periodontal. Con todo ello se hace especial hincapié en la patogénesis de la bolsa periodontal y la reabsorción ósea que son las consecuencias de todo este mecanismo.
Finalizamos con un esquema donde se resumen todos estos factores relacionados con la periodontitis así como una valoración de los factores de riesgo.
PALABRAS CLAVE
Periodontitis, inmunología, interleuquinas, apoptósis, factores de riesgo.
SUMMARY
A review is presented of immunological mechanisms and the destructive process of periodontitis. It begins with the basic concepts of natural and acquired immunity and continues with a study of antigen recognition, inflammation mediators, lines of defence of the organism and risk factors implicated in the disease. This is followed by an exploration of the apoptotic mechanism and an evaluation of the metalloproteinases that are released and are responsible for the periodontal destruction. Special attention is also paid to the pathogenesis of the periodontal pocket and bone reabsorption that are consequences of these mechanisms. Finally, we summarise all of these factors related to periodontitis and provide an assessment of the risk factors.
KEYWORDS
Periodontitis, immunology, interleukins, apoptosis, risk factors.
* Catedrático. Facultad de Odontología. Universidad Complutense de Madrid
** Prof. Titular. Facultad de Odontología. Universidad de Granada
La periodontitis es un proceso inflamatorio producido por ciertas bacterias que tienen una actividad periodontopatógena proveniente de la placa subgingival. Esta interrelación entre las bacterias y los mecanismos de respuesta inmune del hospedador es la base del mecanismo inmunopatológico (1). Las bacterias y sus productos estimulan a las células del hospedador para que liberen ciertos mediadores inflamatorios como las citoquinas y prostanglandinas.
Las enfermedades periodontales constituyen un grupo de enfermedades infecciosas cuyo agente etiológico principal son los microorganismos de la placa bacteriana que se localiza en el surco gingivodental (2,3,4).
Las bacterias anaerobias gram negativas más importantes y prevalentes en el área subgingival son el Actinobacillus actinomycetemcomitans (Aa), Porphlyromonas gingivalis (Pg), Prevotella intermedia (Pi) y Bacteroides forsythus (Bf) (5).
Las bacterias tienen un papel significativo en la patogénesis de la periodontitis participando en la formación de la bolsa periodontal, destrucción del tejido conectivo y reabsorción del hueso alveolar a través de mecanismos directos e indirectos. Sin embargo, la respuesta que se establece es en gran modo responsable del grado de destrucción periodontal así como del balance que se establece entre los diferentes componentes de la misma respuesta (5).
Una vez establecida la periodontitis, se forma un infiltrado inflamatorio constituido por diferentes tipos celulares como linfocitos y macrófagos que van a producir distintos subtipos de citoquinas que participarán en la activación de los procesos de destrucción del tejido conectivo de inserción periodontal (5). Las citoquinas son mediadores biológicos responsables de la inmunopatología de diversas enfermedades (6) de manera que la secreción de ciertos tipos de citoquínas y no de otros, pudieran ser básicos en el desarrollo de una determinada enfermedad.
La migración leucocitaria es básica para eliminar bacterias y centrar las células inmunes en sitios donde la presencia de antígenos es constante y su control depende de acciones asociadas entre moléculas de adhesión, quimioquinas y la expresión de sus receptores. El papel de las distintas quimioquinas y sus receptores en la migración leucocitaria es defendido por Butcher (7) aunque su implicación en la progresión de la periodontitis es de reciente investigación. Aunque gran número de bacterias son capaces de producir una degradación directa de los tejidos, Birkedal - Hasen y cols (8), sugieren que el tejido conjuntivo del hospedador se degrada principalmente por mecanismos del propio hospedador. La pérdida de tejido conjuntivo representa un mecanismo de defensa del hospedador que intenta protegerse merced a la proliferación del epitelio de unión en su sentido apical (9-10).
La manifestación de la enfermedad depende de la interacción entre factores del hospedador, ambientales y del agente microbiológico por lo que es probable que un ambiente específico y factores genéticos sean los que puedan en cierto grado determinar la susceptibilidad del individuo (8, 11). Por lo tanto la microbiota bacteriana periodontopatógena es necesaria pero no suficiente para que exista enfermedad siendo necesario la presencia de un hospedador susceptible (12).
La respuesta inmune también esta regulada por la selección y muerte de las células inmunocompetentes mediante un mecanismo específico de muerte celular conocido como apoptósis (13) siendo este mecanismo de muerte de las células que conforman el infiltrado inflamatorio presente en la periodontitis el factor que participa también en la patogénesis de la enfermedad (14, 15).
La etiología infecciosa de las gingivitis y periodontitis esta ampliamente demostrada, Y así desde el punto de vista que etiopatogénico no existen dudas que estas enfermedades en sus diferentes variantes clínicas, se deben a la placa subgingival y que las bacterias que colonizan esta, son las causantes del proceso destructivo de las estructuras periodontales. Los estudios epidemiológicos han demostrado una asociación significativa entre la gravedad de las enfermedades periodontales la cantidad de placa dental y grado de higiene bucal (16). Los estudios clínicos longitudinales en el hombre señalan que existe una relación causa - efecto entre formación, acumulo de placa dental y desarrollo de gingivitis. El estudio de gingivitis experimental de Lee (17) es taxativo, así como el realizado en animales de experimentación por Lindhe (18). Todos ellos demuestran que el acúmulo de placa siempre es previo al comienzo de la enfermedad. Por otro lado los estudios también confirman que se puede conseguir una prevención y control de la enfermedad tanto por una terapéutica antimicrobiana ( control de la placa y mantenimiento) como con una terapeutica antimicrobiana quimoterápica (19-21).
Referente al potencial patogénico que desarrollan las bacterias no esta claro ya que pudiera ser los productos tóxicos que desarrollan y su posterior entrada en los tejidos el verdadero mecanismo lesivo. Pero ante él tenemos la resistencia del hospedador que va a influenciar el inicio y el desarrollo de la enfermedad. En salud hay un equilibrio entre agresión de bacterias y resistencia del huésped que si se rompe, bien sea por aumento del número y/o virulencia de los gérmenes o bien por una disminución de las defensas, surge la enfermedad.
La enfermedad periodontal es un término general que se refiere a todas las enfermedades que afectan a las estructuras de sostén de los dientes, que afecta a la encía, al ligamento periodontal, cemento radicular, al hueso alveolar y tejidos gingivales. En un principio se limita a encía (gingivitis) progresando por colonización bacteriana de superficies dentales y extendiéndose a tejidos más profundos para destruir la inserción de tejido conectivo al cemento, formándose bolsas, destruyendo hueso alveolar, movilizando los dientes y procediendo a su caída (periodontitis).
Las bacterias al actuar sobre el tejido conectivo provocan una serie de reacciones inflamatorias e inmunológicas en el hospedador (22,23). Durante esta respuesta inflamatoria se presenta una acumulación de células que pueden asociarse a la activación de procesos de destrucción del tejido conectivo (24). Hay estudios longitudinales que sugieren un curso episódico como un modelo que explicaría la progresión de la enfermedad, con episodios de destrucción del tejido periodontal, seguidos de episodios de quietud en el que no se presentan evidencias de destrucción e incluso podría presentarse una cierta regeneración del tejido conectivo perdido (25-27).
Estos episodios de destrucción periodontal se asocian a cambios en la población celular que confirma el infiltrado inflamatorio localizado en el tejido conectivo subepitelial con una disminución importante en la población fibroblástica y un incremento en el número de células inflamatorias principalmente neutrófilos, macrófagos, células plasmáticas y linfocitos en los sitios que muestran actividad (28).
Una vez establacida la periodontitis se forma un infiltrado inflamatorio constituidos por linfocitos T y macrófagos que producen subtipos selectivos de citoquínas que van a participar en la activación de los procesos de destrucción del tejido conectivo de inserción periodontal. Sin embargo las citoquinas son mediadores biológicos responsables de las lesiones inmunopatológicas (29) de tal manera que la liberación de ciertos tipos de citoquinas y no de otras puede ser fundamental en el desarrollo de una determinada enfermedad.
CONCEPTOS BÁSICOS EN INMUNOLOGÍA
EL SISTEMA INMUNE
El sistema inmune es importante en la medida que sus alteraciones funcionales son causa de numerosas enfermedades y en su estudio se imbrican tanto los aspectos anatómicos, como son los órganos y tejidos que lo constituyen, así como el estudio de una serie de células y moléculas liberadas en la respuesta del organismo ante la entrada de un antígeno.
La función principal del sistema inmune es la defensa del organismo frente a agentes patógenos. En el medio ambiente existe una gama amplia de microorganismos que son patógenos para el hombre, el cual no podría vivir sino tuviera mecanismos capaces de atravesar el crecimiento de estos patógenos, una vez que atravesaron la barrera mucocutánea. Los mecanismos son:
1. La inmunidad natural que es la más primitiva, presentándose en animales invertebrados. Esta formada por elementos solubles como proteínas, complemento, citoquinas e interferones. En el campo celular tenemos los mononucleares (monocítos-macrófagos) polimorfonucleares (neutrófilos) con capacidad fagocítica así como las células asesinas naturales (células NK, Natural Killers). Con estas células no hay capacidad de reconocimiento específico del antígeno.
2. La inmunidad adquirida es desde el punto de vista evolutivo más avanzado (se presenta en vertebrados) coexistiendo con la inmunidad natural. Se trata de un sistema complejo que presenta células y moléculas muy específicas para los antígenos. De este sistema son responsables los linfocitos y gracias a ellos la respuesta inmune mejora en los sucesivos contactos con el mismo antígeno en virtud de una cualidad muy importante que es la capacidad de recordar el primer contacto antígeno. Por ello este sistema tiene memoria y especificidad que son dos cualidades que definen la respuesta inmune (figura 1).
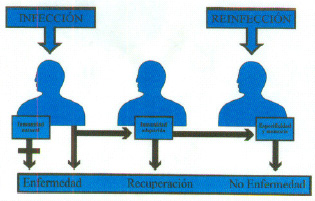
Fig. 1. Inmunidad natural y adquirida
El ejemplo más clásico es la vacunación como medio de prevención de las infecciones, el cual consiste en someter a un sujeto a exposiciones frente a una forma no patógena del agente infeccioso. Después al entrar en el sujeto la forma virulenta del agente, se desarrolla una respuesta inmune muy rápida (memoria) y eficaz (especificidad) que va a eliminar el agente infeccioso antes que se extienda por el organismo.
ESTRUCTURA DEL SISTEMA INMUNE
No esta circunscrito a un solo tejido, sino que es un conjunto de varias estirpes celulares que van a interaccionar entre si bien en forma directa o merced a los productos sintetizados por las células. Estas están distribuidas por todo el cuerpo humano estando en contacto por moléculas del tipo de citoquinas, complejos antígeno-anticuerpo, inmunoglobulinas etc, También pueden estar en contacto directo célula-célula. Los linfocitos actúan como portadores de información (citoquinas). Este sistema va a diferenciar lo propio (constituyente del individuo) de lo ajeno ( no propio o antiguo). El sistema al reconocer un elemento como no propio, va a activar los mecanismos celulares que van a destruir el antígeno. Esto es la respuesta inmune. (figura 2).
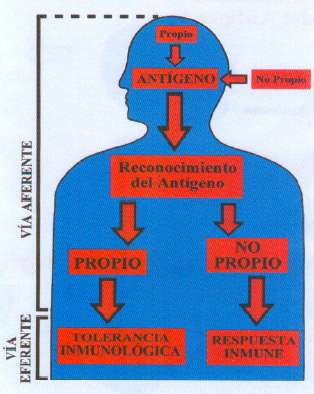
Fig. 2. Reconocimiento del antígeno
FUNCIÓN DEL SISTEMA INMUNE
Este posee dos vías para desarrollar su función la vía aferente y la eferente (figura 2).
La vía aferente reconoce si un elemento debe considerarse como propio o no propio. Si el elemento se reconoce como propio hay un mecanismo de respuesta, es decir, una tolerancia inmunológica, siendo los linfocitos los que aprenden a tener una falta de respuesta para los componentes del propio organismo.
Esto se realiza por un sistema de reconocimiento denominado complejo mayor de histocompatibilidad que en el hombre es el HLA (Human Leucocyte Antigen).
Este sistema diferencia la individualidad entre las especies sino también entre individuos de la misma especie. Si el elemento se reconoce como no propio se pone en marcha un mecanismo de vía eferente de la inmunidad que es la fase de respuesta inmunológica cuya finalidad es destruir el antígeno. Para ello se necesita la fase de presentación del antígeno que facilita que después de la entrada del antígeno en el organismo, este se reconozca como extraño, como no aferente al organismo y se desarrolle la respuesta inmunológica.
RECONOCIMENTO ANTÍGENICO
Cuando un elemento extraño (antígeno) entra en el organismo se produce la captación del antígeno por ciertas células llamadas células presentadoras de antígenos (son principalmente los macrófagos y células dendríticas) que van a fagocitar el elemento extraño (figura 3).
En el interior de la célula se realiza la segunda fase que es el procesamiento del antígeno. Esto se lleva a cabo por las enzimas y la maquinaria celular que transformará el elemento (sea bacteria, virus, sustancia extraña etc.) en elementos más pequeños como proteínas y péptidos. Estos péptidos antigénicos se llevan desde el citoplasma de la célula hasta la membrana plasmática para exponerlos en la superficie celular y ser reconocidos por los linfocitos. Estos péptidos se exponen junto con moléculas HLA en la fase de la presentación del antígeno. En este momento el linfocito capta el antígeno procesado junto las moléculas HLA del individuo que va a permitir a estos linfocitos reconocer lo propio (figura 3).

RESPUESTA INMUNE
A partir de este momento son los linfocitos los que una vez reconocido el antígeno darán lugar a la respuesta inmune siempre que intervengan moléculas de adhesión y las interleuquinas. Los linfocitos no solo poseen en superficie los receptores específicos para el reconocimiento del antígeno, sino que expresan una serie amplia de moléculas coestimuladas, de adhesión y accesorias, que van a facilitar los contactos intercelulares. Estas moléculas intervienen también en la trasmisión de señales al interior de la célula (CD4,CD8,CD2,CD 40 etc.). (figura 4).
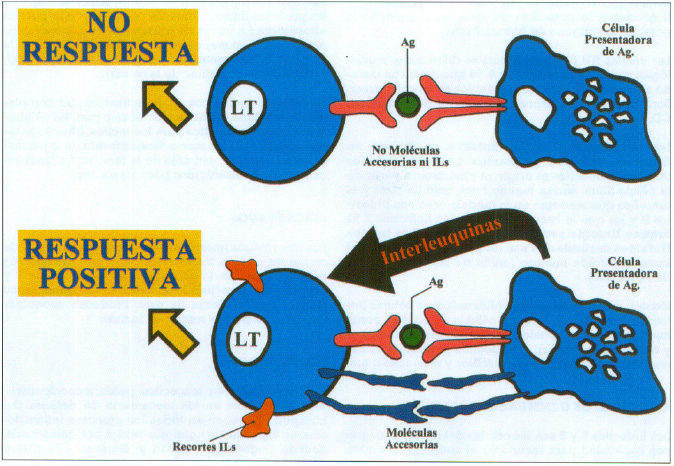
Fig. 4. Respuesta positiva y No respuesta en el reconocimiento antigénico
En la primoinfección, que es el primer contacto con un agente patógeno, la respuesta inmune no es inmediata pudiendo tardar días ya que los linfocitos van a necesitar un tiempo para multiplicarse (proliferación celular) y madurar (diferenciación celular). La diferenciación celular lleva unos linfocitos a la secreción de moléculas específicas contra el antígeno que los estimuló (anticuerpos), a otros al desarrollo de actividad citológica (destrucción celular) y a otros a la liberación de factores solubles que activan a las células con capacidad fagocítica y aumento por lo tanto de la capacidad bacteriana. También los anticuerpos al unirse a los antígenos, estimulan las células fagocíticas así como el complemento. La conexión entre los elementos específicos (linfocitos), y los inespecíficos (células fagocíticas o complemento) son muy importantes en la respuesta inmune.
El sistema inmune tiene una serie de mecanismos reguladores que van a permitir una respuesta adecuada y eficaz cada vez que aparece un antígeno. Estos mecanismos reguladores se intercalan entre la fase de reconocimiento antígenico y la fase de respuesta inmune constituyendo la fase de inmunorregulación. Intervienen también regulando el sistema inmune el nervioso y el endocrino.
CÉLULAS DEL SISTEMA INMUNE
Los linfocitos T y B son las células responsables de la respuesta inmune específica. Los linfocitos B están especializados en la producción de inmunoglobulinas y llevan a cabo la respuesta de anticuerpos frente a un estímulo antigénico (respuesta humoral). Los linfocitos T desarrollan la respuesta mediada por células (respuesta celular). El fenómeno primario es el reconocimiento del antígeno por parte de los linfocitos Ty B gracias a la presencia de receptores específicos para dicho antígeno en su membrana.
Las células NH (Natural Killer) se diferencian morfológicamente de linfocitos Ty B,ya que tienen un tamaño algo mayor y presentan gránulos en su citoplasma. Son linfocitos que expresan una serie de receptores CD.
Los organismos y tejidos linfoides se dividen en órganos linfoides primarios que son aquellos donde los linfocitos se originan y maduran a partir de la célula Stem, serían hígado fetal, médula ósea y el timo. Los que maduran en la médula ósea son linfocitos B y los que lo hacen en el timo los linfocitos T. El órgano linfoide secundario es el bazo. Los tejidos linfoides secundarios son los ganglios linfáticos y el sistema linfoide mucoso (anillo de Waldeyer, Placas de Peyer, etc)
Existen una serie de tipos celulares como células linfoides (linfocitos T, B, NK) y células accesorias como los fagocitos mononucleares (monocítos, macrófagos y células dendríticas), granulocitos poliformonucleares (neutrófilos, esinófilos, basófilos y mastocitos) etc.
ANTICUERPOS O INMUNOGLOBULINAS
Los linfocitos T y B son las células del sistema inmune con capacidad para reconocer el antígeno mediante sus receptores de membrana específicos. Las inmunoglobulinas son glicoproteínas sintetizadas por los linfocitos B y células plasmáticas que tienen la propiedad de unirse de forma específica al antígeno. Todas ellas tienen una estructura básica común dos cadenas pesadas o células H (Heavy) y dos cadenas ligeras o cadenas L (light). Cada cadena pesada se une a una cadena ligera por puentes disulfuro. La molécula Ig adquiere entonces una configuración especial en forma de Y. En función de la cadena pesada se definen una serie distinta de Inmunoglobulinas como Ig G, IgA, IgM, IgD e IgE. Los dos brazos de la Y por separado tienen capacidad de unirse al antígeno.
Las funciones son la de anticuerpo (capacidad de unión específica al antígeno), activación del complemento, atravesar la placenta, unión a ciertas proteínas microbianas etc. La simple unión del anticuerpo al antígeno es suficiente a veces para neutralizar la patogenicidad de un microorganismo o de una toxina microbiana. Otras veces esta unión de antígeno-anticuerpo no es suficiente y se necesitan otros mecanismos adicionales como la activación del complemento, la fagocitosis (opsonización) etc.
El sistema de complemento es un conjunto de proteínas que van a participar en la destrucción tisular y en los procesos inflamatorios. Se puede producir por vía alternativa ( a través de estructuras no dependientes del sistema inmune) o por la vía clásica (mediante la detencción de complejos antígeno-anticuerpo (lgG, IgM) sobre la superficie de la célula).
Los efectos biológicos de la activación del complemento son daño citolítico y citotóxico para las células, actividad quimiotactica para leucocitos, liberación de histamina desde los mastocitos, aumento de la permeabilidad vascular, estímulo de la fagocitosis, inactivación de lipopolisacáridos bacterianos etc.
MACRÓFAGOS
Los macrófagos intervienen en todas las etapas de la respuesta inmune. Intervienen como primera linea de defensa antes de la activación de los linfocitos T y después de fagocitar y procesar el antígeno, como células presentadoras de este. Producen citoquinas importantes para la respuesta inmune.
NEUTRÓFILOS
Se llaman también leucocitos poliformonucleares y son importantes en los mecanismo de defensa del huésped. Aparecen en todos los procesos inflamatorios, en especial los agudos, atraídos por quimiotaxis. Rodean (fagocitosis), matan y digieren a la mayor parte de los microorganismos, neutralizando las sustancia nocivas.
MASTOCITOS
Son importantes por los gránulos citoplasmáticos que contienen histamina, heparina y bradiquinina que se liberan en los tejidos. La desgranulación de los mastocitos se efectúa durante las reacciones inmediatas de hipersensibilidad.
PROTEINAS REGULADORAS
En el sistema inmune existen una serie de factores secretados por los linfocitos T, macrófagos y células aceesorias que lo van a regular. Genéricamente se han denominado citoquinas y van a intervenir en la comunicación entre las células regulando una serie de funciones como son la quimiotaxis, inflamación, inmunidad, reparación tisular etc. Son todos ellos péptidos de bajo peso molecular y se pueden sintetizar por distintos tipos de células, pero las que lo hacen por las células linfoides se denominan linfoquinas o interleuquinas.
En general una interleuquina va a producir más de un efecto (pleitropia). Muchas son factores de crecimiento como la IL-2 para los linfocitos T, la IL-4 para las células B y la IL-5 para los eosinofilos. Van actuar también como factores de diferenciación de leucocitos como la IL-5 que actuará en la diferenciación de células B para que se desarrollen en células productoras de anticuerpos, la IL-6 es factor de diferenciación de linfocitos B y factor de crecimiento para células plasmáticas.
La IL-1 se presenta en forma de IL-1_ e IL-1_, siendo sintetizada en su mayoría por el macrófago y otras células presentadoras de antígeno. Interviene en la secrección de prostangladinas, glucocorticoides y reabsorción ósea.
La IL-2 es el principal factor de proliferación de linfocitos T. La producen las células T cooperadoras (Th-1) después de contactar con el antígeno y recibir señales de la IL-1.
La IL-3 esta más asociada a la diferenciación hermatopoyética.
La IL-4 es responsable del crecimiento de las células B.
La IL-5 esta implicada en la proliferación eosinófila.
La IL-6 esta sintetizada por células Th-2 y células presentadoras de antígeno. Esta implicada en la respuesta inflamatoria.
La IL-7 esta asociada a los procesos de desarrollo del timo y las primeras etapas de la hematopoyesis.
La IL-8 forma parte de la familia quimioquinas y se secreta por los macrófagos siendo quimiotáctica para neutrófilos.
La IL-9 estimula el crecimiento de mastocitos.
La IL-10 es producida por células Th-2, linfocitos B y macrófagos. Actúan sobre las células presentadoras de antígeno inhibiendo su función. Es un regulador negativo de la respuesta inmune celular.
La IL-12 estimula la proliferación de las células T citotóxicas y favorece la activación de células NK.
TNF (_o _) es producida por los macrófagos interviniendo en la actividad osteoclástica y al estimularla puede producir reabsorción ósea. Ayuda a los leucocitos intercambiando su fagocitosis y quimiotaxia.
IFN-_ además de poseer acciones antivirales, es un factor de activación de macrófagos. Interviene en la reabsorción ósea inhibiendo la proliferación y diferenciación de las células primitivas de osteocclastos.
RESPUESTlA INMUNE EN LA PERIODONTITIS
La respuesta inmune ha sido uno de los pilares fundamentales donde se basó el concepto patogénico de la enfermedad periodontal (5).En la década de los años 90 se postuló que las bacterias son esenciales para el inicio de la enfermedad periodontal, pero que existen factores como el tabaquismo y los de índole genética que pueden ser determinantes de la diferente expresión clínica y severidad de los distintos tipos de periodontitis. Las bacterias causan la destrucción tisular activando diversos componentes del sistema inmune del paciente, jugando no solo una función de protección sino también de defensa al mismo tiempo que serán en algún grado responsables de la destrucción en las diferentes etapas inflamatorias de la enfermedad. (figura 5)
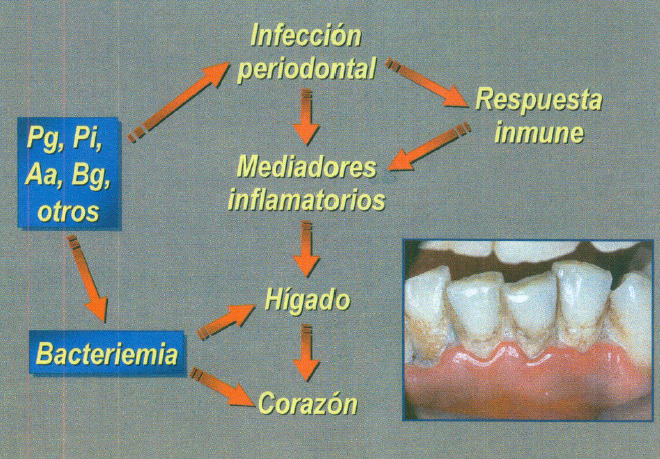
Fig. 5. Periodontopatógenos implicados en la infección
Los mecanismos de actuación de los factores de virulencia son directos por invasión de los tejidos produciendo sustancias nocivas que llevaran a la muerte celular e indirectos por la activación de células inflamatorias que sean capaces de producir y liberar una serie de mediadores que actúan sobre efectores, con una potente actividad proinflamatoria. Todo esto se realiza de una manera secuencial en la que a la microbiota patógena sucede la actuación de los neutrófilos que irán a defender y fagocitar. Si la defensa es adecuada sucederá una gingivitis y una enfermedad limitada, pero si es al contrario habrá una penetración bacteriana que tiene una actuación no solo a nivel sistémico (cardiopulmonar, renal etc.) Sino hacia la periodontitis que pasará por diversas etapas y grados de severidad. Se llega con ello a la actuación del eje linfocito-monocito y a la liberación de los mediadores inflamatorios y citoquinas que destruirán el tejido, expresado clínicamente por la formación de la bolsa periodontal y la pérdida de hueso que a su vez volverán a cerrar el círculo y aumentarán la presencia de la microbiota patógena. En todo este esquema aparecerán los factores de riesgo tanto ambientales como adquiridos (diabetes, tabaquismo) y los factores genéticos. (figura 6).

Fig. 6. Esquema patogénico del proceso destructivo.
Sin embargo, no esta explicado el papel del hospedador en su totalidad así como lo que determina la susceptibilidad, tasa de progresión y el tratamiento y recidiva del proceso. Aquí el genotipo positivo o negativo juega un importante papel en la expresión clínica de la enfermedad (figura 7).

Fig. 7. Relación entre severidad de la enfermedad y el genotipo.
INFILTRADO INFLAMATORIO
En salud el epitelio del surco y de unión evita la invasión bacteriana actuando como una barrera efectiva a la entrada de la mismas o de sus productos. El flujo continuo de limpieza (antoclisis) de la saliva, junto con los anticuerpos y el flujo gingival crevicular actuarán como mecanismos defensivos ante la infección.
La entrada de las bacterias o de sus productos favorece la llegada al tejido conectivo provocando una vasodilatación e inflamación de los vasos sanguíneos. El aumento en el número de neutrófilos que van a migrar a través del epitelio de unión, llegará a formar una parte principal del infiltrado inflamatorio, al mismo tiempo que las células más coronales del epitelio de unión comienzan a proliferar con lo que se va a iniciar la formación de bolsa periodontal. En el infiltrado también existen macrófagos que aumentarán en la evolución de la gingivitis a periodontitis. (figura 8)
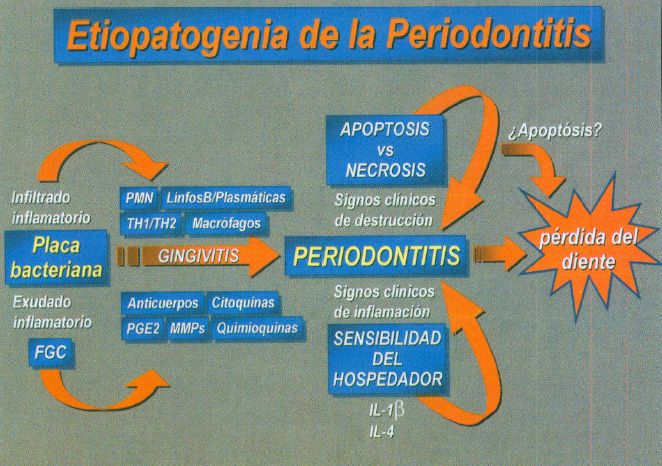
Fig. 8. Etiopatogenia de la periodontitis
Al principio, en los primeros estados, el infiltrado esta dominado por linfocitos T, CD4+, Th 1y Th 2. Después hay un aumento en el número de células B y plasmáticas (30, 31). El movimiento y posterior desplazamiento de los leucocitos desde los vasos hasta los tejidos esta regulado por diferentes tipos de moléculas (32, 33).
Los lipopolisacáridos (LPS) de las bacterias actúan sobre las células como macrófagos, linfocitos, fibroblastos y osteoblastos/osteoclastos.
La proteína de unión al LPS forma un conjunto con este permitiendo su unión a un receptor específico el CD14, presente en los monocitos. La activación del receptor CD14, producirá la secreción de moléculas proinflamatorias como la IL1, IL6, TNF, PGE2 . Estas moléculas a su vez, liberan los mediadores secundarios de la inflamación, como el factor de activación de plaquetas (PAF), bradiquinina e histamina (aminas bioactivas) y las prostaglandinas.
Se ha especulado que la presencia de niveles altos de CD14 en pacientes con enfermedad periodontal al compararse con sujetos sanos podría sugerir una relación con la exposición crónica al LPS (34). El complemento y su activación en forma de cascada con cerca de 30 proteínas tanto en su vía clásica como alternativa tienen como un primer efecto la opsonización de las bacterias por los anticuerpos, posibilitando la destrucción bacteriana. La vía clásica se activa por el complejo antígeno-anticuerpo y la vía alternativa se inicia por LPS u otros productos bacterianos que van a producir la ruptura directa del C3 e iniciar la activación de la cascada.
Las bacterias implicadas en la enfermedad periodontal tienen diferentes mecanismos para evadir al sistema de complemento e incluso algunas poseen una actividad proteolítica en su superficie celular que es capaz de degradar a ciertos componentes del sistema del complemento como C3 y C5. También tienen la capacidad de liberar al medio moléculas capaces de unirse al complemento, de manera que la actividad sobre la bacteria se vea disminuida. Bacterias como Pg y Aa presentan estructuras hidrocarbonadas con gran poder antigénico que desarrolle una respuesta importante de Ig G2.
Los neutrófilos es uno de los mecanismos más importantes y también de los primeros a nivel defensivo local. Estas células salen de los vasos sanguíneos inflamados y migran desde la microcirculación del tejido conectivo gingival hacia el epitelio de unión. En su trayecto realizan funciones de fagocitosis y destrucción bacteriana con lo que al actuar como un mecanismo defensivo impedirán la extensión lateral y apical de la placa.
En esta migración hay dos mecanismos que la regulan, por un lado la expresión de moléculas de adhesión en las células epiteliales y una nueva familia de citoquinas de bajo peso molecular, las quimioquinas, que ejercen atracción sobre diferentes tipos de leucocitos como la IL-8( atrae neutrófilos), MCP-1 (atrae macrófagos) y RANTES (que atrae macrófagos y linfocitos T especialmente del subtipo Th1).
Las moléculas de adhesión intercelular actúan en la interrelación entre leucocitos y queratinocitos expresando receptores tipo integrinas que se encuentran en la superficie de los leucocitos. Las quimioquinas también actúan reclutando y activando en forma selectiva los leucocitos en el lugar de la inflamación.
Algunas bacterias periodontopatógenas PG y Aa son capaces de evadir a los neutrófilos los cuales se desgranularán liberando proteasas de las bacterias, producirán la degradación de la matriz extracelular. En enfermedades en los que los neutrófilos están alterados sea en su cantidad como su cualidad (neutropenia cíclica, síndrome de Chediak-Higashi) o inducidas por drogas (penicilina, femilbutazona etc.) se presenta una alta tasa de destrucción periodontal.
Los anticuerpos son producidos por las bacterias que activan a las células plasmáticas y se presentan como respuesta a la entrada de antígenos bacterianos. Sin embargo los niveles de anticuerpos generados contra las bacterias no se correlacionan significativamente con el estado inflamatorio periodontal.
La destrucción del tejido óseo es en gran parte estimulada por la liberación de prostanglandinas. Se ha visto una asociación positiva entre los niveles de PGE 2 en el fluido crevicular y el aumento de la severidad y agresividad de la enfermedad. El macrófago gingival activado por LPS va a producir altos niveles de PGE 2junto con las citoquínas producidas por macrófagos que también liberan PGE2.
El resultado final es la estimulación de los osteoclastos, la pérdida del tejido conectivo de inserción, el hueso y el tejido conectivo gingival.
La destrucción de la matriz de tejido conectivo se lleva a cabo por las metaloproteinasas que van a ser sintetizadas por monocitos, macrófagos, queratinocitos y células endoteliales. Las MMPs son una familia de proteinasas dependientes de Zn2+ y Ca2+ capaces de degradar la mayoria, sino todas, los componentes de la matriz extracelular. Se van a clasificar en colagenasas intersticiales (MMP-1, 8, 13), gelatinasas (MMP2 y 9), estromalisinas (MMP3, 10, 11), enamelisinas (MMP-20),matrilisinas (MMP-7)y las MMP tipo membrana (MMP 14,15,16 y 17) (35).Laregulación de ellas es en gran medida realizada por IL-1, TNF y Pg. Los patógenos periodontales median la degradación del tejido conectivo durante el desarrollo de la enfermedad periodontal.
Estas enzimas son expresadas en niveles bajos en los tejidos pero pueden sobre-expresarse durante el desarrollo embrionario y durante los procesos de remodelación de los tejidos, en los procesos inflamatorios, durante la invasión tumoral y en las metastasis. Como ocurre con otras proteasas, las MMPs se secretan como pro-formas inactivas que se activan tras digestion proteolítica y que están fuertemente reguladas mediante inhibidores especificos de tejidos (TIMPS).
La regulación de la actividad de las MMPs puede ser a cuatro niveles:
1. Regulación de la transcripción de los genes de las MMPs; además de la regulación específica de cada una de ellas por distintos factores, se ha visto que el LPS, las proteinasas de microorganismo s y las lectinas pueden jugar un papel importante en la regulación de la transcripción de las MMPs. La regulación de muchas MMPs se realiza principalmente en la transcripción, donde la expresión de dichas MMPs es regulada por citoquinas y factores de crecimiento como: IL-1b, IL-la, TNF-a,TGF-b/EGF,PDGF. La IL-1 y el TNF-a están presentes en el FGC en localizaciones inflamadas con concentraciones significativas fisiológicamente (8,36).
2. Activación del precursor: El estado de latencia puede ser mantenido, al menos en parte, por un enlace de coordinación de una Cys con el Zn2+. La ruptura de dicha unión es un pre-requisito para su activación y puede conseguirse mediante distintas vías. Posteriormente la enzima cataliza una autolisis hasta dar lugar a la forma totalmente procesada.
3. Diferencias en la especificidad hacia el sustrato (8): Teóricamente todas son capaces de romper gelatinas y fibronectinas a la misma velocidad y la mayoría rompen colágeno IV y V a altas temperaturas, pero existen especificidades, a grandes rasgos las colagenasas degradan principalmente colágeno fibrilar, las estromalisinas proteoglicanos y glicoproteínas y las gelatinasas colágeno no fibrilar o desnaturalizado.
4. Inhibidores de MMPS (8): Las a-macroglobulinas, particularmente la a2-macroglobulina tienen un papel importante en la regulación de la actividad de las MMPs. Las MMPs activadas son inhibidas por los TIMPs (37,38), que forman complejos bimoleculares mediante uniones no covalentes con la forma activa, pero también en algunas ocasiones con los precursores. LosTIMPs actúan por eliminación de las proteinasas y por bloquear las reacciones de autolisis que dan lugar a su activación. El inhibidor de las metaloproteinasas (TIMP-1) se encuentra en células similares a los macrófagos distribuidos por el tejido y ocasionalmente en células del estroma. El TIMP-1 inhibe, bien por disminución de la actividad de las MMPs, bien por disminución de la activación del precursor a las colagenasas intersticiales y a la MMP-9, mientras que el TIMP-2 es específico de la MMP-2 (39).
Estudios recientes permiten sugerir que la degradación de tejido periodontal solo puede ocurrir en pacientes o en sitios donde el nivel de inhibidores de metaloproteinasas sea bajo, y el nivel de metaloproteinasas sea alto (40). Es posible esperar que un período de destrucción periodontal se encuentre correlacionado con un desbalance entre la expresión de metaloproteinasas y sus inhibidores, y este desbalance biológico podría ser un significativo marcador de destrucción en la periodontitis (39). El asumir que la acumulación de leucocitos pueda contribuir a una mayor producción de MMPs puede explicarnos en parte la destrucción que se produce en la periodontitis y se ha visto que la masiva infiltración inflamatoria, predominantemente de Linfocitos T y Macrófagos, es crítica para el progreso de la enfermedad, ya que liberan citoquinas y producen o estimulan la producción de MMPs por células del entorno. La interacción de estas células inflamatorias contribuye directamente a la sobreproducción local de MMPs (8,36,41).
Las metaloproteinasas son producidas por muchas células bajo el control de factores de crecimiento y citoquinas. El origen celular de las MMPs involucradas en la enfermedad periodontal no está claro, así en el fluido crevicular se ha observado actividad colagenolítica, probablemente del tipo PMN (8,42),aunque cabe la posibilidad, que la actividad de colagenasa de neutrófilos en CGF sea un artefacto debido a la la tisis de PMN durante el procesamiento de las muestras. Otros autores afirman que las MMPs de fibroblastos gingivales, células epitetiales y macrófagos, que responden a citoquinas, factores de crecimiento y varios mediadores inflamatorios son más probablemente las causantes de la degradación de la matriz extracelular durante la progresión de la enfermedad periodontal. (43).
Se ha visto que las citoquinas pueden regular la expresión de MMPs y además estas últimas son capaces de regular la acción de las primeras de forma que MMP-1 y MMP-8 ven incrementada su exptresión con IL-1b, TNF-a, PDGF, TGF-a, EGF, bFGF, NGF y salvo algunas excepciones es anulado ese efecto con TGT-b e INF-g. La expresión de MMP-13 en células del epitelio periodontal es incrementada por TNF-a, TGF- a y TGF-b (44). La MMP-2 en cultivos se expresa constitutivamente y responde de forma moderada a factores de crecimiento como TGT-b e INF-g. En cuanto a la MMP-9 su expresión es inducida por factores de crecimiento: TGT-b e INF-g (45). MMP-3 en células del estroma puede ser constitutiva o puede ser expresada tras la inducción por factores de crecimiento, citoquinas (IL-1, EGF,TNF-a, PDGF,TGF-a, bFGF, NGF) y salvo algunas excepciones es anulado ese efecto con TGT-b e INF-g. El TIMP-1 ve su expresión incrementada por factores de crecimiento y citoquinas (EGF,TNF-a, IL-1 TGF-b, IL-10). Mientras que TIMP-2 ve su expresión disminuida por TGF-b (8). La IL-6 incrementa los TIMPs (46).
Existen estudios en los que aparecen diferencias en la inducción de MMPs respecto a las diferentes citoquinas en función de si se trata de una respuesta Th1 o Th2 (47), así IL-4 incrementando la inducción producida por TNF o contactos célula-célula de la MMP-1, mientras que inhibe la producción de la MMP-9.
MECANISMOS APOPTÓTICOS EN LA PERIODONTITIS
La apoptósis es una muerte celular programada y siendo un proceso fisiológico del desarrollo embrionario y participe de la homeostasis de los tejidos, también es un suceso que acontece en la respuesta inmune. En esta actuación la célula participa activamente en su propio proceso destructivo y siendo conveniente para la supervivencia del organismo se convierte en una respuesta importante en la periodontitis (48). Al entrar la célula en apoptósis se presentan una serie de cambios morfológicos localizados en diferentes estructuras de la misma, como condensación citoplasmática y nuclear y ruptura del ADN en fragmentos internuclosomales. En su estado final la célula presenta una fragmentación en los llamados 11 "cuerpos apoptóticos" que se eliminan por las células fagocíticas sin producir inflamación ni daño celular (48,49). En la necrosis a diferencia de la apoptosis se desencadena una reacción inflamatoria de células y del tejido circundante entre las que se encuentran gran cantidad de enzimas proteolíticas (50). Una de las técnicas que se utiliza para detectar la fragmentación del DNA por las endonucleasas, durante el proceso apoptótico, es la técnica del TUNEL (51,52).
En la membrana plasmática de la célula hay unos receptores que van a regular la apoptósis como Fas (CD95) y Fas ligando (FasL).
FasL puede inducir apoptosis en aquellas células que expresan el receptor Fas. Otro de los mecanismos implicados en la apoptosis celular es la activación de una serie de proteínas citosólicas conocidas como caspasas. La familia de las caspasas son proteasas tipo cisteina que se sintetizan como proenzimas inactivas que son procesadas en las células que sufren apoptosis. (figura 9)
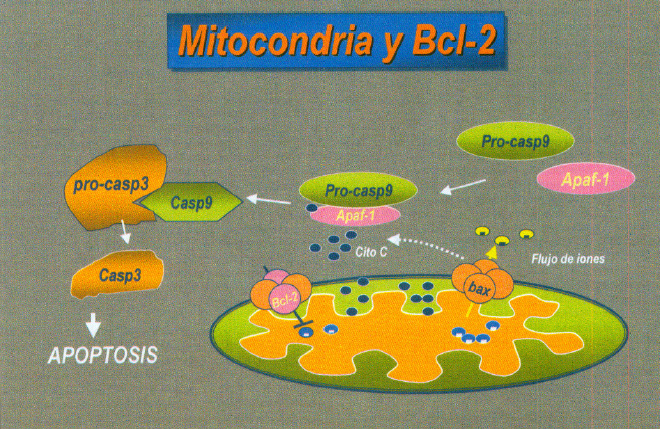
Fig. 9. Mecanismo apoptótico y caspazas.
La caspasa 3 es una proteasa clave que se activa en el proceso de la apoptosis (53, 54). La forma caspasa 3 activa esta presente en las células que estan experimentando apoptosis.
Hoy dia no es posible atribuir la etiología de la periodontitis a un agente bacteriano específico, sin embargo hay una serie de bacterias que van a desarrollar un papel esencial en la iniciación y desarrollo de la enfermedad. La periodontitis en humanos se asocia con anaerobios Gram (-) (55), aunque solo son un grupo entre 10 y 15 los causantes de la enfermedad (56). El nombre de patógeno periodontal se asigna a aquellas bacterias que tienen mecanismos específicos de alteración de los sistemas de defensa del hospedador causando destrucción tisular periodontal (57). Las bacterias asociadas a los diferentes tipos de periodontitis son Aetinobacillus actinomycetemeomitans (Aa), P gingivalis (Pg), B.Forshytus (Bf), P intermedia (Pi), algunas especies de Capnocitophaga, Campilo-bacter restus (Cr), eikerella Couroders (Ec), Espiroquetas, Fusobacterium nucleatum (Fn) y Eubacterium.
En la periodontitis se produce la liberación de endotoxina o lipopolisacáridos (LPS) que inducen la síntesis de citoquinas proinflamatorias por los macrófagos. Estas citoquinas (IL 8, IL 6, IL 1_) aumentan en el fluido crevicular y tejido conectivo gingival de pacientes con periodontitis y actuando en forma activa en la patogénesis de los diferentes tipos de periodontitis (58,59). (figura 10).
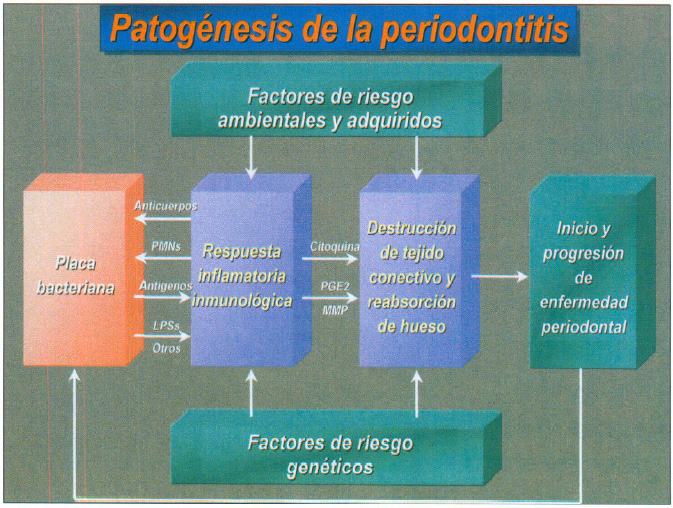
Fig. 10. Patogénesis de la periodontitis.
Así pues los periodontopatógenos liberan antígenos y LPS y el hospedador responde con los PMNs y Anticuerpos. Esta respuesta inmunoinflmatoria se caracteriza por mediadores de la inflamación como son las citoquinas y prostanglandinas que actúan sobre el tejido conectivo y hueso expresándose clínicamente con los signos de la periodontitis. A todo este esquema general se añaden los factores genéticos del hospedador (figura 11).
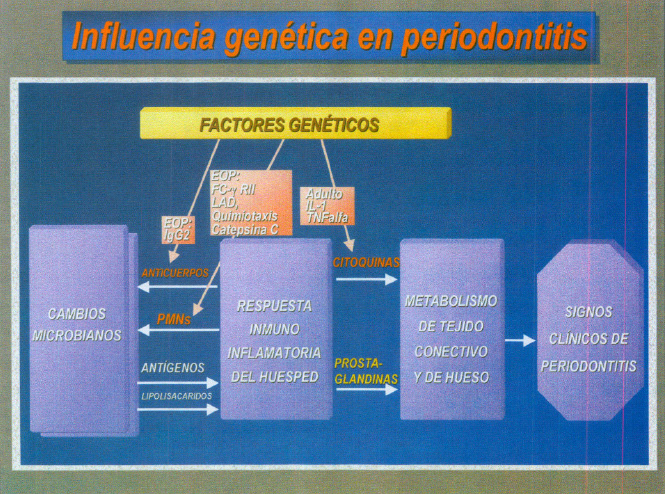
Fig. 11. Influencia genética en la periodontitis.
Junto a estos últimos se añaden factores de riesgo como diabetes, tabaquismo y otros que modularán la enfermedad y su manifestación clínica (figura 12).
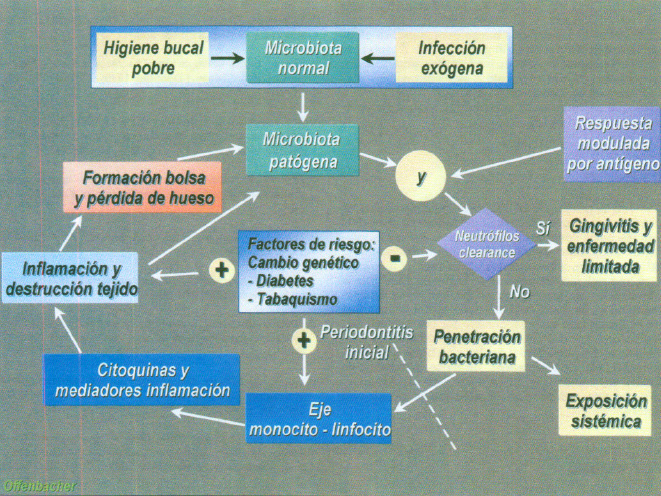
Fig. 12. Esquema parogénico de la destrucción periodontal en relación con los factores de riesgo.
CORRESPONDENCIA
Prof. Antonio Bascones Martínez
Director del Departamento de Medicina y Cirugía Bucofacial
Facultad de Odontología. Universidad Complutense de Madrid
C/ Boix y Morer, 6 1º
28003 Madrid
Telf.: 91 533 42 12
Fax: 91 534 58 60
E-mail: avances@arrakis.es
BIBLIOGRAFÍA
1. Sigusch B., Klinger G. Glokmann E, y Simon H.U. Early - onset and Adult periodontitis associated with abnormal cytokine Production by activated T Lymphocytes. J. Periodont. 1998: 10: 967-78. [ Links ]
2. Ellison SA. Oral bacteria in periodontal disease. J Dent Res 1970; 59 (Suppl 2): 198-202. [ Links ]
3. Socransky, SS. Relationship of bacteria to the etiology of periodontal disease. J Dent Res 1970; 49 ( Suppl 2): 203-22. [ Links ]
4. Socransky, SS. Microbiology of periodontal disease-present status and future considerations. J Periodontol 1977; 48: 497-504. [ Links ]
5. Page RC, Kornman KS.The pathogenesis of human periodonttitis: An introduction. Periodontology 2000.1997; 14: 33-40. [ Links ]
6. Kelso a. Cytokenes in infection desease. Aus Microbiol 1990; 11: 372-6. [ Links ]
7. Butcher EC, Picker LJ.Lymphocyte homing and homeostasis. Science 1996; 272: 60-6. [ Links ]
8. Birkedal Hausen H. Roles of cytokines and inflammatory mediators in tissue destruction. J Periodont Res 1993; 28: 500-10. [ Links ]
9. Genco, RJ.; Evans, RT.; Ellison, Sa. Dental research in microbioly with emphasis on periodontal disease. JADA 1969; 78: 1016-26. [ Links ]
10. Tonetti M, Cortellini D ang Lang N. In situ detection of apoptosis at sites of chronic bacterially induced inflammation in human gingival. Infect Immun 1998; 66: 5190-5. [ Links ]
11. Offenbacher S. Periodontal Diseases: Pathogenesis. Ann Periodontology 1996; 1: 821-78. [ Links ]
12. Seymour G. Importance of the host response in the periodontium. J Clin Periodontol 1991; 18: 421-6. [ Links ]
13. Cohen J, Duke R. Apoptosis and programmed cell death in immunity. Annu Rev Inmunol 1992; 10: 267-93. [ Links ]
14. Sawa T,Nishimura F,Ohyama H. et al. In vitro induction of activation-induced cell death in lymphocytes from chronic periodontal lesions by exogenous Fas ligand. Infect Immun 1999; 67: 1460-4. [ Links ]
15. Koulouri O, Lappin Df, Radvar M, Kinane DF. Cell division, synthetic capacity and apoptosis analysed by in situ hybridization and immunohistochemistry. J Clin Periodontol 1999; 57: 1605-13. [ Links ]
16.Waerhaug J. healing of dento -epithelial junction following the use of dental floss. J Clin periodontal 1981: 8: 166-50. [ Links ]
17. Löe H, Theilade E, Jensen SB. Experimental gingivitis in man .J. Periodontol 1965; 36: 177-87. [ Links ]
18. Lindhe J, Hamp SE, Löe H. Experimental periodontitis in the Beagle dog.J Periodontal Res 1973; 8: 1-10. [ Links ]
19. Ramfjord SP.Longitudinal Study of periodontal therapy. J Periodontal 1973; 44: 66-77. [ Links ]
20. Axelsson P, Lindhe J. Effect of controlled oral procedure on caries an periodontal disease in adults. J Clin Periodontal 1978; 8: 133-51. [ Links ]
21. Loesche WJ. Chemotherapy of dental plaque infections. Oral Sci Rev 1976; 9: 65-72. [ Links ]
22. Kornman K, Page RC; Tonetti M, Periodontology 2000. 1997; 14: 33-40. [ Links ]
23. Page RC, Korman KS.The pathogenesis of human periodontitis: An introduction. Periodontology 2000.1997; 14: 9-11. [ Links ]
24. Lindhe J, Haffajee A, D., socransky S. Progresion of periodontal disease in adult subjects in the absence of periodontal therapy, j Clin Periodontol 1983; 10: 433-42. [ Links ]
25. Goodson JM, Tanner, A.C.R., Haffajee A.D., Sornbenger G.C, Socransky S.S. Patterns of progression and regression of advanced destructive periodontal disease. J Clin Periodontol 1982; 9: 472-81. [ Links ]
26. Haffajee A, Socransky S, Goodson J. Clinical parameters as predictor of destructive periodontal disease activity. J Clin Periodontol 1983; 10: 257-95. [ Links ]
27. Haffajee A.D., Socransky S.S., and Goodson IM. Comparision of different data analyses for detecting changes in attachment level. J Clin Periodontol 1983; 10: 298-310. [ Links ]
28. Zappa U, Reinking-Zappa M, Graf H, Espeland M. Cell populations and episodic periodontal attachment loss in humans.J Clin Periodontol 1991; 18: 508-15. [ Links ]
29. Kelso A. Cytokines in infection desease. Aus Microbiol 1990; 11: 372-6. [ Links ]
30. Zappa U, Reinking-Zappa M, Graf H, Espeland M. Cell populations and episodic periodontal attachment loss in humans. J Clin Periodontol 1991; 18: 508-15. [ Links ]
31. Saglie F, Pertuiset J, Rezende M, Nester M, Marfany A, Cheng J. In situ correlative inmuno-identification of mononuclear infiltrates and invasive bacteria in diseased gingival.J Periodontol 1988; 69: 688-96. [ Links ]
32. Osborn L.Leukocyte adhesion to endothelium in inflammation. Cell 1990; 62: 3-6. [ Links ]
33. Zimmerman G, Prescott S, McIntyre T. endothelial cell interactions with granulocytes: tethering and signaling molecules. Inmunol Today 1992; 13: 93-100. [ Links ]
34. HayashiJ, Masaka T, Ishikawa I.Increased levels of soluble CD14 in sera of periodontitis patients. Infet Inmun 199; 67 (1): 417-20. [ Links ]
35. Woessner, J. (1998). "The matrix metalloproteinase family", Academic Press, San Diego. [ Links ]
36. Birkedal-Hansen, H., Moore, W. G., Bodden, M. K., Windsor, L. J., Birkedal-Hansen, B., DeCarlo, A., and Engler, J.A. Matrix metalloproteinases: a review. Crit Rev Oral Biol Med. 1993; 4 (2): 197-250. Review. [ Links ]
37. Ryan, M. E., Ramamurthy, S., and Golub, L. M. Matrix metalloproteinases and their inhibition in periodontal treatment.Curr Opin Periodontol. 1996; 3: 85-96. Review. [ Links ]
38. Shibata, Y., Takiguchi, H., and Abiko, Y. Antisense oligonucleotide of tissue inhibitor of metalloproteinase-l induces the plasminogen activator activity in periodontal ligament cells.J Periodontol. 1999 Oct;70(10): 1168-65. [ Links ]
39. Lacraz, S., Nicold, L. P., and Dayer, J. M. IL-10 inhibits metalloproteinase and stimulates TIMP-1 production in human mononuclear phagocytes. J Clin Invest. 1996 Nov; 96 ( 5): 2304-10. [ Links ]
40. Reynolds, J. J., and Meikle, R. C. Mechanisms of connective tissue matrix destruction in periodontitis.Periodontol 2000.1997 Jun; 14: 144-57. Review [ Links ]
41. Ding, Y., Uitto, V. J., Firth, J., Salo, T., Haapasalo, M., Konttinen, Y. T., and Sorsa, T. Modulation of host matrix metalloproteinases by bacterial virulence factors relevant in human periodontal diseases.Oral Dis. 1995 Dec; 1 (4): 279-86. [ Links ]
42. Birkedal-Hansen H. Role of cytokines and inflammatory mediators in tissue destruction. J. Periodont. Res. 1993; 28: 500-10. [ Links ]
43. Golub, L. M., Sorsa, T., Lee, H.-M., Ciancio, S., Sorbi, D., Ramamurthy, N. S., Gruber, B., Salo, T., and Konttinen, Y.T. Doxycycline inhibits neutrophil (PMN)-type matrix metalloproteinases in human adult periodontitis gingiva. J Clin Periodontol.1995; 22 (2): 100-9. [ Links ]
44. Murphy, G., and Gavrilovic, J. Proteolysis and cell migration: creating a path? Curr Opin Cell Biol. 1999 Oct; 11 (5): 614-21. Review. [ Links ]
45. Suzuki, J., Isobe, M., Kawauchi, M., Endoh, M., Amano, J., and Takamoto, S. Altered expression of matrix metalloproteinases and tissue inhibitors of metalloproteinases in acutely rejected myocardium and coronary arteriosclerosis in cardiac allografts of nonhuman primates. Transpl Int. 2000; 13 (2): 106-13. [ Links ]
46. Lacraz, S., Isler, P.,Vey, E., Welgus, H. G., and Dayer, J.-M. Direct contact between T lymphocytes and monocytes is a major pathway for induction of metalloproteinase expression. J Biol Chem. 1994 Sep 2; 269 (35): 22027-33. [ Links ]
47. Chizzolini, C., Rezzonico, R., de Luca, C., and Burguer, D. D.,J-M. Th2 cell membrane factors in association with IL-4 enhance matrix metalloproteinase-l (MMP-1) while decreasing MMP-9 production by granulocyte-macrophage colony-stimulating factor-differentiated human monocytes. J Immunol. 2000 Jun 1;164 (11): 5952-60. [ Links ]
48. Kerr J, Wyllie A, Currie A. Apoptosis: a basic biologic phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972; 26: 239-57. [ Links ]
49. Abrams J,Whitee K,Fessler L,Steller H. Programmed cell death during Drosophila embryogenesis. Development 1993; 177: 29-43. [ Links ]
50. Majno G and Joris J. Apoptosis, oncosis and necrosis. An overview of cell death. Am J Pathol 1995; 146: 3-16. [ Links ]
51. Enari M, Sakahira H,Yokoyama H, Okawa A, Iwamatsu A, Nagata S. A caspaseactivated Dnase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998; 391: 45-50. [ Links ]
52. Sakahira H, Enari M,Nagata S.Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 1998; 391: 96-9. [ Links ]
53. Nicholson D, Ali A, Thornberry N, Vaillancourt J, ding C, et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995; 376: 37-43. [ Links ]
54. Pantel T, Gores G, Kaufmann S. The role of proteases durig apoptosis. FASEB J 1996; 10: 587-597. [ Links ]
55. Clark W,Löe H. Mechanism of initiation and progression of periodontal disease. Periodontology 2000.1993; 2: 72-82. [ Links ]
56. Haffajee Ad, Socransky SS, Smithe J, Dibart S. Relation of baseline microbial parameters to future periodontal attachment loss. J Clin Periodontol 1991; 18: 744-750. [ Links ]
57. Carlsson J. What is a Periodontal Pathogen? Periodontology Today. 1988 Int. Congr., Zurich, pp. 104-111. [ Links ]
58. Masada M, Persson R, Kenney J, Lee S, Page R, Allison A. Measurement of interleukin 1_ and -_in gingival crevicular fluid: Implication for the pathogenesis of periodontal disease. J Periodont Res 1990; 25: 156-63. [ Links ]
59. Tsai C, HO Y and Chen C. Levels of interleukin -1_ and interleukin -8 in gingival crevicular fluids in adult periodontitis. J Periodontol 1995; 66: 853-9. [ Links ]

