










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkINTRODUCCIÓN
La amiloidosis primaria (AL) es la forma más común de amiloidosis (71 %)1, que comprende un grupo de enfermedades caracterizadas por el depósito en diversos tejidos de una sustancia precursora de la sustancia amiloidea. Presenta una población clonal de células plasmáticas que producen una cadena ligera monoclonal de tipo lambda o kappa. Su tasa de incidencia en Estados Unidos se estima en 6-10 casos / 1.000.000 habitantes / año con una edad media al diagnóstico entre los 60 y 70 años. Junto con la sospecha clínica (proteinuria o síndrome nefrótico, miocardiopatía restrictiva y hepatomegalia entre otras) se precisa de la confirmación histológica y la caracterización del tipo de amiloide para establecer el diagnóstico definitivo y la elección del tratamiento (trasplante autólogo o quimioterapia) 2.
El mieloma múltiple (MM) es una enfermedad neoplásica caracterizada por la proliferación de células plasmáticas en médula ósea y por la sobreproducción de inmunoglobulinas monoclonales completas. Es la segunda neoplasia hematológica en orden de frecuencia y las lesiones óseas, la hipercalcemia, la insuficiencia renal, la anemia y elevación de la velocidad de sedimentación globular (VSG) son las principales alteraciones clínicas. La introducción de nuevos fármacos junto con el avance en las medidas de soporte (diálisis, bisfosfonatos, vertebroplastias, etc.) han mejorado la supervivencia de los pacientes3.
Presentamos el caso de una paciente con AL asociada a MM con características únicas en su desarrollo.
CASO CLÍNICO
Mujer de 68 años con antecedentes personales de psoriasis ungueal (diagnosticada por servicio de Dermatología) e intervenida de síndrome del túnel del carpo. Acude a nuestra consulta de Atención Primaria por disnea de esfuerzo progresiva desde hace tres meses tras un cuadro de bronquitis crónica, ortopnea de dos almohadas y ocasionalmente episodios presincopales en decúbito supino relacionados con el esfuerzo junto con dolor torácico de características atípicas. En la anamnesis dirigida comenta también que desde hace aproximadamente tres años presenta hematomas periorbitarios y supraclaviculares autolimitados que aparecen y desaparecen sin causa desencadenante.
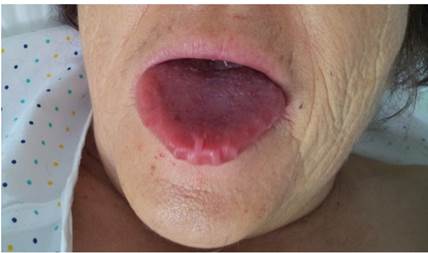
En la exploración física destacaba un regular estado general, leve ingurgitación venosa yugular, eupneica en reposo con saturación de oxígeno del 94 % basal, afebril, macroglosia (Figura 1) y edemas maleolares leves. En la auscultación cardiaca presentaba un soplo sistólico polifocal III/VI y en la auscultación pulmonar se apreciaban crepitantes bibasales. A nivel cutáneo se detectó la presencia de hematomas periorbitarios (Figura 2) y supraclaviculares, así como la destrucción de la lámina ungueal en los dedos de ambas manos (Figura 3). La anamnesis y exploración clínica nos sugería un cuadro compatible con insuficiencia cardiaca en el cual había que valorar que fuera primaria o secundaria a endocrinopatías (hipotiroidismo), déficit de vitamina B12, procesos autoinmunes (lupus-like) y neoplasias hematológicas.


Los resultados obtenidos en las pruebas complementarias fueron: hemograma y coagulación normal, VSG de 101 mm, función renal normal, PCR 7,5 mg/dl, NT-ProBNP 4550 ng/l, marcadores biológicos normales, autoinmunidad negativa, perfil tiroideo, ácido fólico y vitamina B12 normales; proteinograma con componente monoclonal en región beta de 1120 mg/dl; electrocardiograma con complejos de bajo voltaje sin otras alteraciones y radiografía de tórax con derrame pleural bilateral leve.
Con el diagnóstico de insuficiencia cardiaca y gammapatía monoclonal a estudio se derivó a la paciente al servicio de Medicina Interna, en donde se realizaron las siguientes pruebas complementarias: 1) Ecocardiografía transtorácica con contraste, que mostró insuficiencia mitral leve, hipertrofia ventricular izquierda concéntrica de predominio septal con imagen radiolucente en su interior (“sparkling”). Función sistólica preservada. Disfunción diastólica moderada-severa. Aurícula izquierda no dilatada. 2) Ergometría convencional normal. 3) TAC torácico que tan sólo mostró derrame pleural de moderada cuantía sin otras alteraciones relevantes.
Ante las evidencias clínicas y en las exploraciones complementarias sugestivas de amiloidosis se realizó una biopsia de la grasa subcutánea que confirmó la presencia de depósito amiloideo con tinción de rojo Congo positiva. Además, se realizó aspirado de médula ósea en base al resultado del proteinograma y alteración de inmunoglobulinas, el cual contabilizó hasta un 50 % de células plasmáticas (típicas y atípicas) con cariotipo 55XX y tipo IgA-lambda compatible con mieloma múltiple, asociado a mal pronóstico según el cariotipo. Se completó el estudio con una serie ósea sin evidencia de alteraciones líticas.
DISCUSIÓN
La incidencia de la AL varía de unos países a otros, siendo en Estados Unidos y el Reino Unido de 6-10 casos por 1.000.000 personas, suponiendo la primera causa de amiloidosis en países desarrollados. La incidencia anual del MM es de 5 casos por cada 100.000 habitantes-año y la prevalencia de AL en pacientes con MM es aproximadamente del 20-35 %1,2. El mieloma múltiple representa el 10 al 15 % de todas las neoplasias hematológicas, siendo más frecuente en la 7ª década de la vida sin diferencias significativas entre ambos sexos3 4-5. La incidencia estimada actualmente en España es de 3,5 casos por cada 100.000 habitantes3,4.
La AL puede afectar a cualquier órgano del cuerpo excepto el cerebro. La sintomatología inicial dependerá de los órganos afectados, siendo la debilidad o fatiga y la pérdida de peso los síntomas iniciales más frecuentes1.
La afectación cardiaca es responsable de hasta el 40 % de mortalidad de los pacientes con amiloidosis sistémica. La insuficiencia cardiaca congestiva suele ser progresiva y de rápida instauración; otras manifestaciones menos frecuentes son las arritmias, la angina, infarto o hipotensión, todo ello consecuencia del depósito del material amiloideo principalmente en miocardio, aunque también puede afectar a endocardio y pericardio. La infiltración miocárdica de material amiloideo se traduce en el ecocardiograma en una imagen patognomónica consistente en un granulado brillante (“granular sparkling”) en la pared del ventrículo izquierdo y septo. En nuestro caso además había datos de infiltración amioloidótica avanzada con importante acúmulo mencionado en septo y cavidades ventriculares aumentadas bilateralmente, pero con ambas aurículas de tamaño normal de forma un tanto sorpresiva. El electrocardiograma presenta característicamente un patrón de bajo voltaje1,2.
La insuficiencia renal se da en grados variables, afecta hasta el 50 % de los pacientes, siendo frecuente la proteinuria, que se manifestará como un síndrome nefrótico con edema e hipoalbuminemia secundaria1,2,6. Entre las manifestaciones gastrointestinales destaca la macroglosia con indentaciones en los bordes laterales, está presente en el 10 al 19 % de pacientes con AL y en el 32 % de la asociada a MM, el depósito de material amiloideo en las glándulas salivales producirá xerosis7. Otros hallazgos menos frecuentes son la saciedad precoz, malabsorción, perforación intestinal, ulceraciones, hemorragia digestiva, hepatopatía1,2.
El compromiso del sistema nervioso periférico, puede producir una neuropatía periférica sensitiva como el síndrome del túnel del carpo, presente en una cuarta parte de los pacientes, o una neuropatía periférica autónoma que se manifestará como hipotensión ortostática, diarrea, etc. 1,2.
Las alteraciones mucocutáneas se dan aproximadamente en el 21 al 40 % de los casos y frecuentemente serán el primer signo de la enfermedad. Lesiones como petequias, placas purpúricas y equimosis se localizan preferentemente en párpados (ojos de mapache), reborde periorbitario, cuello, región submamaria y genitales y se producen espontáneamente o favorecidos por traumatismos mínimos o maniobras de Valsalva; esto podría estar en relación con la fragilidad capilar y coagulopatía consecuencia del depósito amiloideo en los mismos. La onicodistrofia aunque es poco frecuente es muy sugestiva de amiloidosis asociada a mieloma múltiple. Otros hallazgos menos característicos son las placas, pápulas y nódulos, de aspecto céreo, localizados en parpados, cuello, axilas, ingles, genitales, cara y labios1,2.
Nuestra paciente presentó inicialmente disnea como síntoma de afectación cardiaca, equimosis periorbitarias y supraclaviculares, además de onicodistrofia y también neuropatía periférica (síndrome del túnel del carpo), macroglosia y afectación renal con proteinuria en rango no nefrótico6,7.
La estrecha colaboración con el servicio de Medicina Interna permitió, a través de una citación preferente, agilizar las pruebas complementarias que orientaron la resolución del caso clínico.
El diagnostico de confirmación de AL se basa en la demostración del depósito amiloideo, Rojo Congo positiva en la grasa subcutánea (sensibilidad 57-85% y especificidad 92-100 %), medula ósea, biopsia rectal (sensibilidad 84 %) o del órgano presumiblemente afecto. En todo paciente con diagnóstico de AL debe investigarse la presencia de un MM debido a las características clínicas y analíticas que comparten ambas entidades, además de la probabilidad de concurrencia de ambas a la vez5. La supervivencia media de AL en un estudio de 868 pacientes fue de 3,8 años, la mortalidad durante el primer año del diagnóstico alcanzó el 27 % y el 75 % de la mortalidad se debió a fallo cardiaco. Características ecocardiográficas de amiloidosis cardiaca como engrosamiento de pared, disfunción diastólica y reducción de la función sistólica han sido asociadas con un mal pronóstico1. Por lo tanto, el determinante principal de mortalidad es el compromiso cardiaco.
La amiloidosis con o sin mieloma tiene un mal pronóstico, con una media de vida de 12 a 15 meses posteriores al diagnóstico1,5.
En el tratamiento tanto de la AL como del MM el estado general y la edad son determinantes a la hora de discutir las opciones terapéuticas (la quimioterapia y el trasplante de medula ósea autólogo son las opciones terapéuticas más frecuentes para ambas entidades). El tratamiento de soporte estará dirigido a mantener la función de órganos y tejidos afectos (hemodiálisis, eritropoyetina, etc.), aunque debe evitarse el uso de digoxina, bloqueantes beta, calcio antagonistas y IECA si existiese compromiso cardiaco8.
En el caso de nuestra paciente se inició tratamiento con bortezomib-melfalan-prednisona (bisemanal). A los dos meses consecutivos al diagnóstico presentó ingreso hospitalario por shock séptico secundario a Staphylococus aureus meticilin sensible y por dolor torácico típico con shock cardiogénico con elevación muy importante de los biomarcadores cardiacos (Troponina I y NTproBNP). A las 4 semanas tras el último alta hospitalaria acude nuevamente por shock cardiogénico y fallo multiorgánico, produciéndose finalmente el fallecimiento.
En conclusión, la insuficiencia cardiaca es una patología muy prevalente en nuestras consultas de Atención Primaria por lo cual es preciso un despistaje de las causas secundarias que pueden llegar a provocarlo, realizando un amplio diagnóstico diferencial. Es necesario un manejo multidisciplinario gracias a la continuidad asistencial entre Atención Primaria y Hospitalaria. El diagnóstico precoz es esencial en la AL, antes de que el daño ya se haya establecido, y siempre investigar la posibilidad de un MM asociado dada la frecuente concurrencia de ambos.

