










 Servicios personalizados
Servicios personalizados
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google 
 Permalink
PermalinkIntroducción
La policitemia vera (PV) es una neoplasia mieloproliferativa (NMP) crónica que resulta de la expansión clonal de un progenitor hematopoyético y que se distingue de otras NMP por la eritrocitosis. El 99% de los pacientes presentan una mutación somática en los exones 12 o 14 del gen Janus kinasa 2 (JAK2) y la gran mayoría son portadores del alelo JAK2 V617F1,2.
La clasificación oficial de la Organización Mundial de la Salud (OMS) de los tumores de tejidos hematopoyéticos y linfoides de 2016, establece los criterios diagnósticos para las siete subcategorías de NMP relacionadas con mutaciones JAK2/CARL/MPL, incluyendo PV, trombocitemia esencial y mielofibrosis primaria. Esta clasificación introduce una serie de cambios con respecto a la anterior de 2008, entre los que destaca la disminución del valor umbral de hemoglobina y hematocrito, así como el concepto de PV enmascarada, lo que supone un cambio en el panorama diagnóstico y terapéutico de este subtipo de síndromes mieloproliferativos y revela que la PV ha sido infradiagnosticada en el pasado. La OMS considera la combinación de aspectos clínicos, morfológicos y de genética molecular como la forma más adecuada para identificar este tipo de enfermedades3.
El inicio de la enfermedad suele ser insidioso, con manifestaciones relacionadas con el aumento de la viscosidad de la sangre causado por el aumento del componente celular que conduce a falta de oxigenación, lo que provoca síntomas que incluyen cefalea, trastornos visuales, vértigo y tinnitus. Alrededor del 20% de los pacientes pueden presentar episodios de trombosis arterial o venosa que, en ocasiones, puede ser la primera manifestación de la enfermedad. Los hallazgos clínicos son inespecíficos, pudiendo incluir visceromegalia, plétora facial o nódulos gotosos4.
Se han identificado diversos factores de riesgo y genéticos predisponentes, como la edad avanzada y los antecedentes trombóticos. Los niveles elevados de leucocitos y la mutación de JAK2 superior al 50% se han relacionado con peor evolución1,4.
El tratamiento de la PV tiene como objetivo reducir las complicaciones, mejorando así la supervivencia. Los eventos tromboembólicos son la principal causa de morbilidad y mortalidad, por lo que su prevención constituye el objetivo principal de cualquier estrategia terapéutica2. La piedra angular del tratamiento de la PV consiste en flebotomías periódicas para mantener el hematocrito por debajo del 45% y en la utilización de bajas dosis de ácido acetilsalicílico. En los pacientes de alto riesgo entre 40 y 75 años, se recomienda la utilización de hidroxicarbamida como tratamiento de primera línea e interferón α o anagrelide como segunda línea1.
Diagnóstico de osteomielitis mediante pruebas de imagen
La osteomielitis o infección ósea puede estar relacionada con una lesión penetrante o con la diseminación de la infección desde tejidos adyacentes o vía sanguínea. A diferencia de la celulitis, suele requerir tratamiento antibiótico a largo plazo y posiblemente cirugía, por lo que es importante un diagnóstico temprano para un establecer un tratamiento adecuado. La gammagrafía ósea en tres fases con 99mTc-difosfonatos es una prueba de imagen sensible y precisa, que permite diferenciar entre osteomielitis y celulitis al poner de manifiesto el remodelado óseo secundario a la osteomielitis antes de que se produzcan cambios anatómicos visibles en las imágenes radiográficas. Por su baja especificidad, requiere de pruebas complementarias para confirmar el diagnóstico. En este sentido, la gammagrafía con leucocitos marcados con 99mTc-exametazima es de elección, al posibilitar un diagnóstico más rápido que otros radiofármacos gracias a su mayor disponibilidad. Los anticuerpos monoclonales antigranulocitos marcados con 99mTc son una buena alternativa, si bien presentan el inconveniente de una posible respuesta inmune cruzada5,6,7.
Caso Clínico
Varón de 49 años, fumador, intervenido de fractura de tobillo derecho hace más de 15 años que presentaba una zona ulcerosa en la cara medial del astrágalo-calcáneo con mala evolución y que fue remitido a Medicina Nuclear para valorar un posible proceso infeccioso asociado.
En el estudio gammagráfico óseo con 99mTc-difosfonatos en tres fases (vascular ósea, precoz y tardía) se observó la fijación del radiofármaco en zona de articulación tibio-astragalina, con intenso componente osteoblástico, así como en zona adyacente del cuboides (Figura 1), por lo que se programó un estudio con leucocitos marcados con 99mTc-exametazima.

Figura 1: Gammagrafía ósea en tres fases con 99mTc-difosfonatos. (A) Imágenes dinámicas; (B) Imagen precoz; (C) Imagen tardía
Según los Procedimientos Radiofarmacéuticos de la Real Farmacopea Española8, el marcaje de leucocitos con 99mTc-exametazima requiere su separación previa de los hematíes mediante sedimentación, utilizando hidroxietilalmidón al 6% para facilitar la separación celular. En estas condiciones, el tiempo de sedimentación puede variar de 30 a 75 minutos en función de la velocidad de sedimentación globular de cada paciente. En el presente caso no se produjo sedimentación de los hematíes, a pesar de prolongar el proceso durante 120 minutos. Al no poder separar los hematíes de los leucocitos por el procedimiento recomendado, el complejo 99mTc-hexametazima marcaría también los hematíes haciendo que las imágenes no fueran resolutivas9.
Ante la imposibilidad de realizar la prueba, se programó un estudio con anticuerpos antigranulocitos marcados con 99mTc que no mostró anomalías en la fijación del radiofármaco indicativas de un proceso infeccioso (Figura 2).
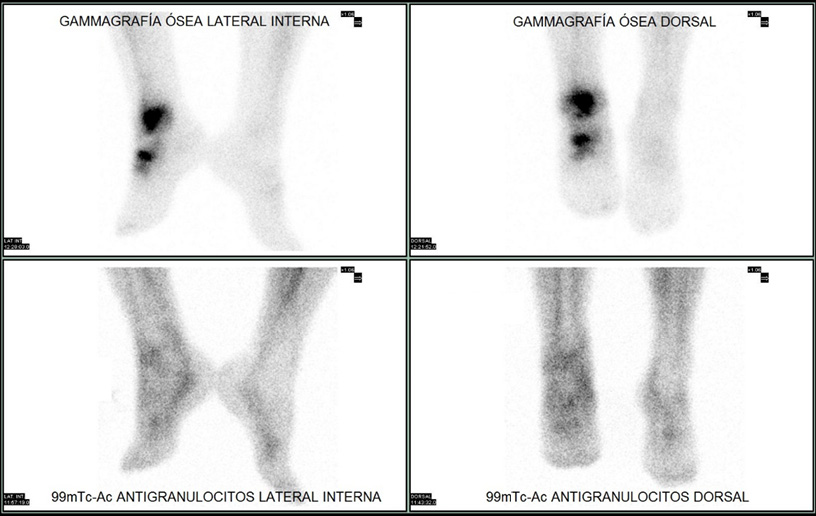
Figura 2: Estudio comparativo entre imágenes tardías de gammagrafía ósea con 99mTc-difosfonatos (arriba) que muestra hipercaptación del radiofármaco e imágenes de gammagrafía con anticuerpos antigranulocitos marcados con 99mTc (abajo) en las que no se observan anomalías
Existen diversas situaciones que pueden ocasionar disminución de la velocidad de sedimentación globular (VSG), llegando a ser de 0 mm, entre las que destacan los síndromes de hiperviscosidad, las poliglobulias, el hábito tabáquico, la insuficiencia cardíaca y la leucocitosis extrema10.
Aunque el hecho de ser fumador podría explicar la baja VSG, el aspecto rubicundo que presentaba el paciente durante la extracción de sangre indujo a consultar el hemograma en busca de posibles alteraciones. Se observaron valores aumentados en todas las series sanguíneas: leucocitos (13,49x109/L), neutrófilos (9,61x109/L), hematíes (7,03x1012/L), hemoglobina (20,8 g/dl), hematocrito (61%), plaquetas (446x109/L) y plaquetocrito (0,49%).
A la vista de estos resultados, se recomendó realizar estudio y valoración hematológica del paciente, que reveló unos niveles séricos de eritropoyetina bajos (1,3 U/L), la ausencia de datos en el frotis sanguíneo sugestivos de existencia de fibrosis medular y la presencia de la mutación V617F del gen JAK2 (4%). El paciente refirió alteraciones visuales ocasionales, mostrando la exploración física facies pletórica y ausencia de esplenomegalia. De acuerdo con los hallazgos, el paciente fue diagnosticado de PV iniciando tratamiento con flebotomías semanales y ácido acetilsalicílico a dosis bajas. Asimismo, se recomendó estricto control de todos los factores de riesgo cardiovascular (tabaquismo, hipertensión arterial y dislipemia) debido a la asociación de la enfermedad con el incremento del riesgo trombótico. Posteriormente, el paciente fue tratado con hidroxicarbamida por la imposibilidad de acudir con frecuencia a las flebotomías.
Conclusiones
En el caso descrito, el paciente presentaba plétora facial, alteraciones visuales, poliglobulia y factores de riesgo, como hábito tabáquico e hiperlipemia, todo ello concordante con el patrón de la PV. La colaboración del especialista en Radiofarmacia permitió diagnosticar un caso de PV que no había sido identificado hasta ese momento, pese a que el paciente presentaba síntomas desde hacía años.

