










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkAnales de Medicina Interna
versão impressa ISSN 0212-7199
An. Med. Interna (Madrid) vol.22 no.1 Jan. 2005
| Linfomatosis intravascular. Descripción de tres casos M. J. MENÉNDEZ CALDERÓN, M. E. SEGUÍ RIESCO1, M. ARGÜELLES1, J. NUÑO MATEO Servicios de Medicina Interna y 1Anatomía Patológica. Hospital de Cabueñes. Gijón, Asturias
|
| INTRAVASCULAR LYMPHOMATOSIS. A REPORT OF THREE CASES
| ||
| RESUMEN La linfomatosis intravascular (LIV) es una neoplasia poco frecuente caracterizada por la proliferación de células linfoides atípicas en la luz de pequeños vasos (vénulas, capilares y arteriolas). La sintomatología está causada por fénomenos trombóticos debido a la oclusión de los vasos por células tumorales. Los órganos más afectados son el sistema nervioso y la piel, aunque puede estar involucrado cualquier otro sistema, sin afectación de los tejidos linfoides. Presentamos tres casos de LIV: el primero se trata de un varón de 57 años que debuta con demencia y síntomas neurológicos; un segundo caso en un paciente de 69 años con manifestaciones predominantemente pulmonares (tos, disnea y fiebre) y una mujer con fiebre de origen desconocido (FOD) y síndrome inflamatorio sistémico. Dada la ausencia de signos específicos de esta entidad multiorgánica con evolución rápidamente progresiva, su diagnóstico es postmorten en todos nuestros casos. Para su confirmación se precisó de biopsia de uno de los órganos afectados. PALABRAS CLAVE: Linfomatosis intravascular. Linfoma angiotrópico. | ABSTRACT Intravascular lymphomatosis (IVL) is a rare malignancy characterized by a proliferation of atypical lymphoid cells occluding small blood vessels (venules, capillaries and small arteries). The symptoms are caused by embolims due to massive proliferation. Nervous system and skin are the most common sites of involvement but all organs may be involved, although it is typified by the absence of malignant cells in lymphoid tissues. We describe three cases of IVL: first patient was a 57 years old man with rapidly progressive dementia and neurological involvements and second case of a 69 years old man hospitalised with predominant symptoms in the lung (cough, dyspnea and fever), and a woman presenting as fever of unknown origin (FUO) with systemic inflammatory response syndrome. In all cases that we reported have been diagnosed post-morten because of rapid progression of a multisystem disease and the absence of pathognomonic clinical manifestations. Diagnosis can be made using biopsy of one of the involved organs. KEY WORDS: Intravascular lymphomatosis. Angiotropic lymphoma. |
Menéndez Calderón MJ, Seguí Riesco ME, Argüelles M, Nuño Mateo J. Linfomatosis intravascular. Descripción de tres casos. An Med Interna (Madrid) 2005; 22: 31-34.
Trabajo aceptado: 28 de julio de 2004
Correspondencia: María José Menéndez Calderón. Servicio de Medicina Interna. Camino de Cabueñes, s/n. Hospital de Cabueñes, Gijón. Asturias. e-mail: mjmenendezcalderon@yahoo.es
INTRODUCCIÓN
La linfomatosis intravascular o linfoma angiotrópico es una neoplasia hematológica muy rara, caracterizada por la proliferación intravascular de células malignas en el interior de los vasos sanguíneos de pequeño tamaño (1). La sintomatología clínica es la debida a la oclusión vascular, con afectación multiorgánica y de curso rapidamente progresivo. El diagnóstico en la mayoría de los casos es postmorten, dada la infrecuencia y difícil sospecha inicial de este tumor (1,2).
A continuación presentamos tres casos diagnosticados de linfomatosis intravascular tras su confirmación por necropsia.
CASOS APORTADOS
Caso 1: Varón de 57 años, con antecedentes de tabaquismo, etilismo, HTA y ulcus duodenal que ingresa en 1998 por síndrome confusional agudo acompañado de cambios en el carácter y fiebre de 4 meses de evolución. A su ingreso desorientado, hemodinamicamente inestable, Tª 39,5 ºC, siendo el resto de la exploración física normal y sin signos de focalidad neurológica.
En los estudios complementarios destacaba: Hb 9 g/dl, velocidad de sedimentación glomerular (VSG) 50 mmHg 1ª hora, proteína C reactiva (PCR) 284 mg/l, albúmina 1,9 g/l, ALT 62 U/l, AST 54 U/L, GGT 102 U/L, FA 101 U/L, LDH (lactato deshidrogenasa) 1.498 U/L y test de coombs directo positivo.
El análisis de líquido cefalorraquídeo (LCR) era normal y los estudios microbiológicos (cultivos y serologías) fueron negativos. Las técnica de imagen con tomografía axial computarizada (TAC) toracoabdominopélvico, craneal y ecocardiograma fueron normales. Durante su hospitalización presenta deterioro progresivo del nivel de conciencia, artritis de rodillas, pancitopenia, síndrome hemafagocítico, disfunción multiorgánica severa y éxitus.
En el estudio necrópsico: las lesiones macroscópicas fueron enfisema pulmonar con antracosis, hiperplasia nodular de tiroides y arterioesclerosis generalizada. A nivel del sistema nervioso central presentaba infartos isquémicos múltiples (Fig. 1).

En el estudio microscópico destacaba la ocupación de las luces de vasos de pequeño y mediano calibre por células redondas, atípicas de escaso citoplasma y nucleo redondo con nucleolos prominentes y mitosis, que se identificaron como linfocitos por técnicas de inmunhistoquímica. Los órganos más afectados fueron pulmón, piel, cerebro, próstata y testículo.
Caso 2: Varón de 69 años de edad, con antecedentes de tabaquismo, hipercolesterolemia y cardiopatía isquemia tipo angor, que ingresa en el Servicio de Medicina Interna (MI) en el 2002 por clínica de tos, disnea de mínimos esfuerzos, ortopnea, dolor en hemitórax izquierdo, malestar general y edemas periféricos de 2 meses de evolución.
En la exploración física presentaba TA 100/60 mmHg, frecuencia respiratoria de 22/min con ruidos respiratorios disminuidos a la auscultación pulmonar y signos de mala perfusión periférica; no presentaba adenopatías y el resto de la exploración era normal.
En la analítica a su ingreso presentaba una gasometría arterial basal: pO2 49 mmHg y pCO2 27 mmHg, sodio en sangre 128 mEq/L, GGT 110 U/L, FA 156 U/L, LDH 1.133 U/L y albúmina 1.7 g/l. La radiografía de tórax mostraba un pequeño derrame pleural; se realizó toracocentesis diagnóstica: células 85/ml, glucosa 156 mg/dl, proteínas 1,30 g/dl, ADA 16,3 U/L y LDH 29,1 U/L. El TAC de torax y ecocardiograma era normales.
El diagnóstico inicial es de bronquitis aguda y se pautó tratamiento antibiótico, oxigenoterapia y broncodilatadores inhalados. La evolución clínica es desfavorable con progresivo deterioro general con disminución del nivel de conciencia, hipotensión y cuadro agudo de dolor abdominal con hematemesis que provoca su fallecimiento.
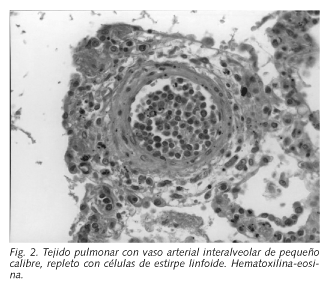
En la necropsia las lesiones macroscópicas encontradas fueron bronconeumonía con derrame pleural y antracosis, arterioesclerosis generalizada y bazo supernumerario. No se realizó examen craneal. El estudio histológico mostró la proliferación de células neoplásicas dispuestas en grupos, no cohesivas a nivel intravascular con mayor afectación en pulmón, piel, tiroides, próstata, riñón y glándulas suprarrenales (Fig. 2 ).
Caso 3: Mujer de 75 años, sin antecedentes patológicos de interés, en seguimiento en consulta de MI desde hace un año por lesiones cutaneas maculopapulosas, recurrentes en miembros, así como anemia acompañado de linfopenia, LDH elevado y anticuerpos antinucleares (ANA) y anticuerpos antifosfolípido (ACL) positivos. Ingresa en el año 2002 para estudio por síndrome constitucional y fiebre intermitente de 4 meses de evolución.
En la exploración física presentaba Tº 38 ºC, TA 110/60 mmHg, lesiones tróficas de carácter crónico en miembros, sin adenopatías, esplenomegalia y el resto de la exploración era normal. En los estudios complementarios destacaban: Hb 9,4 g/dl, leucocitos 2.500/mm3 con 580 linfocitos, VSG 90 mmHg 1ª hora, PCR 51 mg/l, ALT 74 U/L, AST 80 U/L, FA 521 U/L y LDH 3.592 U/L. Se repitió el estudio inmunológico: anticuerpos ANA patrón centrómero 1/960, ACL positivos a 21,70 y determinaciones de complemento, factor reumatoide (FR) y anticuerpos DNA, ENA y ANCA negativos.
Los cultivos de sangre, orina y serologías (hepatitis, lúes, borrelia, brucella y coxiella) fueron también negativos. La exploración ORL, ginecológica, radiografía de tórax, ecografia abdominal, TAC toracoabdominal, ecocardiograma, gammagrafía tiroidea, mamografía y electromiograma fueron normales. Durante su hospitalización presenta deterioro general progresivo con desorientación, edemas, hipotensión y dolor abdominal agudo.
En el control analítico destacaba: hiponatremia severa, hiperpotasemia, acidosis metabólica, elevación de pruebas de función hepática e insuficiencia renal. Su evolución fue desfavorable siendo éxitus.
En el estudio de la necropsia las alteraciones macroscópicas no fueron significativas: hiperplasia de tiroides, discreta dilatación de la vía biliar, ascitis de 250 ml y diverticulosis colónica. A nivel microscópico se observaba obstrucción intravascular de los pequeños vasos por células de estirpe linfoide. El estudio inmunuhistoquímico reveló que dichas células eran positivas para el antígeno leucocitario común y PAN-B (CD-20), confirmando su línea B (Fig. 3). Los órganos con mayor afectación fueron pulmón, piel, tiroides, riñón, suprarrenales y útero.

DISCUSIÓN
La linfomatosis intravascular o linfoma angiotropo es un tumor maligno infrecuente caracterizado por la proliferación de células malignas en la luz de los vasos sanguíneos de pequeño calibre (vénulas, capilares y arteriolas) a nivel multisistémico (2).
Fue descrita por Pfleger y Tappeiner en 1959 con el nombre de angioendoteliomatosis maligna. Inicialmente se creía que tenía un origen neoplásico endotelial, pues la células tumorales eran positivas para el factor VIII (marcador de células endoteliales) que posteriormente se demostró que era falsa (3). Sin embargo, las técnicas inmunohistoquímicas han permitido confirmar el origen linfoide de dichas células neoplásicas; se trata pues de un tipo de linfoma no hodgkin, siendo la mayoría de los casos descritos de estirpe linfoide B (2).
Clinicamente suele manifestarse con síndrome general, fiebre y afectación orgánica difusa de evolución rapidamente progresiva, con expresión clínica variable según la localización de las múltiples microtrombosis. Los órganos más frecuentemente afectados son el sistema nervioso central y la piel (1,4). Las manifestaciones clínicas iniciales más frecuentes suelen ser neurológicas como síndrome confusional, demencia de progresión rápida, infartos isquémicos de repetición o afectación medular, como ocurrió en los casos primero y tercero3,5. Las lesiones cutáneas le siguen en orden de frecuencia y son muy variadas (máculas, nódulos, petequias, etc.). El caso 2 presentó un debút clínico infrecuente con síntomas respiratorios e hipoxemia progresiva con estudios radiológicos normales. Las manifestaciones pulmonares no suelen ser frecuentes, si bien cuando hay repercusión pulmonar clínica presentan un patrón radiológico patológico con afectación intersticial difusa. También se han descrito alteraciones hematológicas, insuficiencia renal y suprarrenal o síndrome de secreción inadecuada de ADH6,4. Por tanto, se pueden afectar múltiples órganos: riñón, pancreas, suprarrenales, corazón, tracto gastrointestinal, genital; no suele haber afectación de ganglios línfáticos, médula ósea y ocasionalmente existe repercusión hepático o esplénica (2,4).
A nivel analítico hay una respuesta inflamatoria intensa con VSG, PCR elevadas y fenómenos de autoinmunidad (anemia hemolítica, leucopenia, ANA ACL, FR, test de coombs positivo). También destacar los valores de LDH muy elevados y alteración de enzimas hepáticos, hiponatremia, hipoalbulinemia e insuficiencia renal. El estudio de LCR no suele mostrar signos patognomónicos ni células malignas (1,3). Las pruebas de imagen son normales, pues la afectación sólo ocurre en los espacios intravasculares sin la formación de masa tumorales (3,5).
Histologicamente se caracteriza por la proliferación de células mononucleares atípicas, pleomórficas, con prominencia nucleolar y actividad mitótica que ocluyen la luz de los vasos de pequeño calibre. Es rara su localización extravascular y son frecuentes la formación de microtrombosis (4,5). Las técnicas inmunohitoquímicas han permitido identificar su naturaleza linfoide, con positividad para los marcadores CD45 (Antígeno Leucocitario Común), CD20 (Marcador Pan-B) y F-VIII negativo, (marcador de células endoteliales) (7). Los órganos con mayor afectación en nuestra experiencia fueron pulmón, piel, tracto genital, tiroides y glándulas adrenales (Tabla I).

Debido a que las manifestaciones clínicas son muy variadas, su sospecha es muy difícil, siendo en la mayoría de los casos el diagnóstico postmorten. La biopsia de los órganos afectos como la piel, aunque no muestren signos macroscópicos de afectación, puede ser de elección para diagnóstico, al estar frecuentemente infiltrada y debido a su fácil accesibilidad5. El diagnóstico diferencial debe hacerse con vasculitis, neoplasias ocultas, fiebre de origen desconocido, demencia o infartos de repetición, infecciones o síntomas pulmonares de etiología no filiada (3,4).
Se trata de una entidad linfoproliferativa rara, con progresión clínica muy rápida y de mal pronóstico, a pesar de utilizar tratamiento con quimioterapia (CHOP: ciclofosfamida, doxorrubicina, vincristina y prednisona), radioterapia, plasmaféresis y corticoides (4,5).
Como conclusión, creemos que debería sospecharse ante un síndrome general, fiebre, afectación cutánea o neurológica, con sintomatología multiorgánica, inflamatoria y acompañados de valores muy elevados de LDH (6). En estos casos deberíamos plantearnos realizar una biopsia de piel que incluya tejido celular subcutáneo, aunque el paciente no presente lesiones cutáneas, para conseguir un diagnóstico precoz.
Bibliografía
1. Lui P, Wong G, Poon W, Tse G. Intravascular lymphomatosis. J Clin Pathol 2003; 56: 468-70. [ Links ]
2. Ip M, Chan k, Chan I. Systemic inflammatory response syndrome in intravascular lymphomatosis. Intensive Care Med 1997; 23: 783-6. [ Links ]
3. Arboix A, Costa I, Besses C. Intravascular lyphomatosis: a rare etiology of recurrent cerebral ischemia. Rev Neurol 2000; 324: 1188-90. [ Links ]
4. Demirer T, Dail D, Aboulafia D. Four varied cases of intravascular lymphomatosis and a literature review. Cancer 1994; 73:1738-1745. [ Links ]
5. Baumann T, Hurwitz N, Karamitopolou-Diamantis E, Probst A, Herrmann R, Steck A. Diagnosis and treatment of intravascular lymphomatosis. Arch Neurol 2000; 57: 374-7. [ Links ]
6. Walls J, Hong Y, Cox J, McCabe K, O'Brien K, Allerton J, Derdak S. Pulmonary intravascular lymphomatosis: presentation with dyspnea and air trapping. Chest 1999; 115: 1207-1210. [ Links ]
7. Murase T, Nakamura S, Tashiro K, Suchi T, Hiraga J, Haysaki N, Kimura M, Murakami M, Mizoguchi Y, Suzuki T, Saito H. Malignant histiocytosis-like B-cell lymphoma, a distinct pathologic variant of intravascular lymphomatosis: a report of five cases and review of the literature. Br J Haematol 1997; 99: 656-664. [ Links ]

