










Meu SciELO
 Serviços customizados
Serviços customizadosServiços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkArchivos de la Sociedad Española de Oftalmología
versão impressa ISSN 0365-6691
Arch Soc Esp Oftalmol vol.82 no.5 Mai. 2007
Coriorretinopatía lacunar como presentación de Síndrome de Aicardi en el lactante
Pathognomonic chorioretinal lacunar lesions in an infant with Aicardis Syndrome
Puertas-Bordallo D.1, Lozano-Vázquez M.2, De Domingo-Barón B.2, Ruiz-Falcó Rojas M.L.3, González-Gutiérrez-Solana L.3, Fernández-Fernández J.2
Sección de Oftalmología/Estrabología del Hospital Infantil Universitario Niño Jesús de Madrid. España.
1 Doctor en Medicina.
2 Licenciado en Medicina.
3 Licenciado en Medicina. Sección de Neurología.
Dirección para correspondencia
RESUMEN
Caso clínico: Se presenta el caso de una niña de 2 meses y 3 semanas de vida que ingresa desde Urgencias por crisis comiciales. Fue diagnosticada en la semana 20 de gestación de agenesia de cuerpo calloso, posteriormente confirmada en ecografía cerebral postnatal. En la exploración del fondo de ojo, se observaron lagunas retinianas (coriorretinopatia lacunar) peripapilares en «sacabocados» en ambos ojos. La resonancia magnética craneal muestra agenesia completa del cuerpo calloso.
Discusión: Las lesiones atróficas coriorretinianas con alteración del epitelio pigmentario de la retina, son fundamentales en el diagnóstico del síndrome de Aicardi.
Palabras clave: Coriorretinitis lacunar, epilepsia, agenesia de cuerpo calloso, espasmos infantiles, síndrome de Aicardi.
ABSTRACT
Clinical case: We report the case of an 81-day-old female infant who was brought to the Emergency Department because of a seizure. At 20 weeks of gestational age she was diagnosed to have agenesis of the corpus callosum, with this being confirmed later by magnetic resonance imaging. Ophthalmological examination of the fundus showed peripapillar chorioretinal lesions (lacunar chorioretinopathy) in both eyes.
Discussion: Chorioretinal lacunar and retinal pigment epithelial abnormalities are the basis for the diagnosis of this syndrome (Arch Soc Esp Oftalmol 2007; 82: 311-314).
Key words: Lacunar chorioretinopathy, epilepsy, agenesis of the corpus callosum, infantile spasms, Aicardi syndrome.
Introducción
El síndrome de Aicardi es un trastorno genético dominante ligado al cromosoma X. Fue descrito por primera vez en 1965 por el neurólogo Jean Aicardi1. Es muy difícil determinar el número exacto de niños con Aicardi, pero se estima que en el mundo se han descrito entre 300 y 500 casos.
Se define por la triada típica (1):
1. Anomalías oftalmológicas (la más específica es la coriorretinopatía lacunar).
2. Agenesia de cuerpo calloso (parcial o completa).
3. Espasmos infantiles.
La mayoría de los casos se diagnostican antes de los cinco meses de vida. Son niñas con un desarrollo normal hasta los tres meses de vida, momento en el que comienzan con espasmos infantiles, que son frecuentemente asimétricos, y que están precedidos por una convulsión tónica o clónica limitada a la zona donde predominan los espasmos. Otras alteraciones neurológicas que se pueden observar son hemiparesia, microcefalia, retrasos motor y mental. En la mayor parte de las neonatas el electroencefalograma (EEG) muestra asincronía y supresión de descargas en las primeras semanas de la vida, lo que indica la ausencia de interconexión entre los hemisferios.
El hallazgo oftalmoscópico más específico es la coriorretinitis lacunar más o menos extensa uni o bilateral. En un 50% de los casos se observa un coloboma del disco óptico. Otras alteraciones oculares que se pueden presentar son microftalmía, depósitos de pigmento retiniano, hipertelorismo, ausencia de reflejos pupilares, estrabismos o persistencia de membranas pupilares.
La resonancia magnética cerebral es la prueba de imagen más informativa y muestra en la mayoría de los casos malformaciones a nivel del sistema nervioso central (SNC) que acompañan a la agenesia del cuerpo calloso.
La existencia de la tríada clásica es diagnóstica de este síndrome en la mayor parte de las ocasiones, sin embargo en los casos raros, sobre todo si no se observa agenesia del cuerpo calloso, el hallazgo de al menos dos de los nuevos criterios mayores (tabla I) sería suficiente para el diagnóstico (2).

Caso clínico
Se presenta el caso de una niña nacida a término. El embarazo tuvo un curso normal con resultado citogenético normal en la amniocentesis realizada a las 16 semanas de gestación. En la semana 20 de la misma se diagnostica de agenesia de cuerpo calloso, posteriormente confirmada en ecografía cerebral postnatal.
A los dos meses y tres semanas de vida la niña es ingresada desde Urgencias por presentar crisis comiciales consistentes en espasmos asimétricos con flexión de tronco y cabeza, flexión o extensión de miembros inferiores, giro de la cabeza y desviación ocular conjugada hacia arriba y hacia la derecha de unos cuatro segundos de duración y que se repiten tras intervalos de 6-8 segundos en forma de salvas de hasta 90 espasmos.
Hemograma, coagulación y bioquímica sérica general normal. El EEG muestra, al ingreso, un registro asimétrico con actividad irritativa y peor organización sobre hemisferio derecho.
Se realizó una valoración oftalmológica que puso en evidencia la existencia de coriorretinitis de aspecto lacunar bilateral (imágenes en «sacabocado» adyacentes a la papila). El resto del estudio oftalmológico no mostró ningún hallazgo patológico.
Ante estos hallazgos clínicos, se sospechó un síndrome de Aicardi por lo que se solicitó una resonancia magnética cerebral que mostró una agenesia completa del cuerpo calloso (figs. 1 y 2) con anomalías asociadas: heterotopía de la sustancia gris del ventrículo lateral derecho (figs. 3 y 4), polimicrogiria y quiste de plexo coroideo en el atrio del ventrículo lateral derecho (fig. 5).
La radiografía costovertebral no mostraba anormalidades.

Fig. 1: Ausencia del cuerpo calloso.

Fig. 2: Ausencia del cuerpo calloso y astas ventrículos
laterales evertidos. Corte coronal T2.
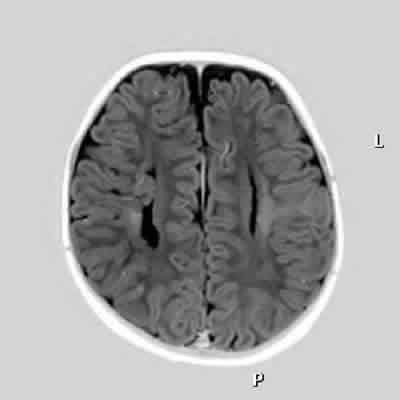
Fig. 3: Heterotopía de la sustancia gris en el ventrículo lateral derecho.

Fig. 4: Alargamiento de los ventrículos laterales por ausencia
del cuerpo calloso. Heterotopía de la sustancia gris del ventrículo lateral derecho.

Fig. 5: Quiste de líquido cefalorraquídeo. Ventrículos evertidos.
Discusión
El síndrome de Aicardi se caracteriza por una asociación de anomalías oftalmológicas, crisis epilépticas (manifestación que se aprecia más precozmente) de difícil control terapéutico (3) o espasmos infantiles y agenesia parcial o total del cuerpo calloso. Su incidencia y prevalencia son bajas y se presenta en el sexo femenino (es letal en varones y sólo se ha descrito en niños con trisomía 46 XXY).
Dentro de las manifestaciones oftalmológicas son las lagunas redondeadas de atrofia coriorretiniana el signo fundamental en el diagnóstico funduscópico (fig. 6). También pueden observarse grandes papilas colobomatosas o estafilomas asociados. Menos frecuentemente existe un estrabismo asociado, microftalmía, membranas pupilares persistentes e hipertelorismo... (4), considerándose todos ellos hallazgos menores a la hora de establecer un diagnóstico certero (tabla I).

Fig. 6: Focos de coriorretinitis lacunar peripapilares.
La oftalmoscopía binocular indirecta en determinadas enfermedades neuropediátricas resulta de gran valor. El estudio funduscópico permite detectar ciertas enfermedades infantiles evitando en muchos casos la realización de otras pruebas diagnósticas complejas, con un coste elevado de las mismas, y una orientación más precisa de la patología que presenta el paciente.
Otras manifestaciones clínicas que pueden presentar estos pacientes son las anomalías vertebrales, una hipotonía generalizada, la existencia de telangiectasias, deformidades de la cabeza y torácicas, asimetrías faciales (5), hemangiomas cutáneos, hipoplasia del quinto dedo, orejas de implantación baja o retraso psicomotor que limita de forma importante la actividad de estos enfermos.
No existe un tratamiento curativo. El síndrome de Aicardi tiene un pronóstico neuroevolutivo muy desfavorable, aunque algunos casos pueden presentar una afectación intelectual de grado moderado.
Bibliografía
1. Aicardi J, Lefebre J, Lerique-Koechlin A. A new síndrome: Spasm in flexion, callosal agenesis, ocular abnormalities. Electroencephalogr Clin Neurophysiol 1965; 19: 609-610. [ Links ]
2. Aicardi J. Aicardi Síndrome: old and new finding. Int Pediatr 1999; 14: 5-8. [ Links ]
3. Prats Vinas JM, Martinez Gonzalez MJ, Garcia Ribes A, Martinez Gonzalez S, Martinez Fernandez R. Callosal agenesis, chorioretinal lacunae, absence of infantile spasms, and normal development: Aicardi syndrome without epilepsy?. Dev Med Child Neurol 2005; 47: 419-420. [ Links ]
4. Lorenzo Rabanal J, Gallego Villalobos M, Puertas Bordallo D, Pérez Belmonte L, Rozas Reyes P, Señaris González A, et al. Síndrome de Aicardi adulto. Diagnóstico oftalmológico. Acta Estrabológica 2001; 1: 13-16. [ Links ]
5. Sutton VR, Hopkins BJ, Eble TN, Gambhir N, Lewis RA, Van den Veyver IB. Facial and physical features of Aicardi syndrome: infants to teenagers. Am J Med Genet A 2005; 138: 254-258. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Diego Puertas Bordallo
Sección de Oftalmología/Estrabología. Hospital Infantil Universitario Niño Jesús
Avenida Menéndez Pelayo, 65
28009 Madrid
España
E-mail: dpuertas.hnj@salud.madrid.org
Recibido: 21/3/06.
Aceptado: 26/3/07.

