










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos Españoles de Urología (Ed. impresa)
versión impresa ISSN 0004-0614
Arch. Esp. Urol. vol.60 no.7 sep. 2007
Enfermedad renal poliquística autonómica recesiva tipo infantil en dos recién nacidos consecutivos de la misma madre.
Infantile type renal autosomal recessive policystic disease in two successive newborns from the same mother.
Francisco Javier Torres Gómez, Antonio Silva Abad y Gloria Reina Vinardell1.
Servicios de Anatomía Patológica y de Ginecología1. Hospital de Jerez de la Frontera. Cádiz. España.
Dirección para correspondencia
RESUMEN
Objetivo: La enfermedad renal poliquística renal autonómica recesiva es responsable de un elevado número de fallecimientos de aquellos que la padecen (formas prenatal y neonatal principalmente).
Métodos: Presentamos el caso de los estudios necrópsicos de dos hijos sucesivos de la misma madre portadores de dicha enfermedad.
Resultados: Analizamos las características macro y microscópicas de esta entidad quística.
Conclusiones: Aquellos pacientes que logran superar la lactancia seguramente sufrirán la complicaciones derivadas de la patología hepática asociada.
Palabras clave: Autosómico recesiva. Enfermedad renal poliquística.
SUMMARY
Objective: Renal autosomal recessive policystic disease is responsible for a great number of deaths among affected individuals (mainly prenatal and neonatal forms).
Methods: We report the necropsy studies of two successive newborns from the same mother with such disease.
Results: We analyze the macroscopic and microscopic characteristics of this cystic disease.
Conclusions: Those children that make it through the breast-feeding period will surely suffer from the associated liver pathology.
Key words: Autosomal recessive. Polycystic renal disease.
Introducción
Si bien la enfermedad renal autosómica recesiva es de sobra conocida, resulta curioso observar su presentación en dos hijos consecutivos de la misma madre sobre todo hoy día en que las técnicas de imagen están lo suficientemente desarrolladas como para asesorar a los progenitores en vista a tomar precozmente la actitud más adecuada al caso previa al fallecimiento del pequeño. Aprovechamos la ocasión para ilustrar los aspectos macro y microscópicos más relevantes de esta patología.
Caso clínico
Dos recién nacidos, varón y hembra de una misma madre y fallecidos a los 10 y 45 minutos de vida respectivamente a los que se les realizó examen necrópsico. El primero de los cadáveres, correspondiente a la hembra, fue remitido con el juicio clínico de insuficiencia respiratoria grave con sospecha de Síndrome de Potter con la constatación de oligoamnios severo; nació mediante cesárea urgente por presentación de nalgas y el test de Apgar fue 1/3/7; minutos más tarde falleció. El examen externo permitió observar una tonalidad subcianótica, facies triangular con hendiduras parpebrales mongoloides, micrognatia, raiz nasal ancha y occipucio prominente. El abdomen, globuloso, duro y ligeramente abollonado permitía la palpación de dos grandes masas ocupando ambas fosas renales y hemiabdomenes. A la apertura de cavidades destacaba la presencia de dos grandes masas renales de 10 x 8 x 5,5 cm y 12 x 8 x 6 cm con pesos de 190 y 235 gr respectivamente. Si bien se podía discernir la silueta renal, la superficie, abollonada, presentaba numerosas formaciones quísticas de contenido seroso; al corte dichos quistes mostraban un tamaño heterogéneo siendo mayores los situados a nivel cortical, dando al riñón un aspecto de esponja. Los pulmones derecho e izquierdo pesaban 17 y 15 gr (peso habitual del conjunto de 49 gr) mostrando una tonalidad rojiza uniforme; ambos se encontraban comprimidos como consecuencia de la elevación diafragmática condicionada por el gran tamaño de los riñones. El resto de los órganos no mostraba alteraciones macroscópicas significativas salvo las alteraciones posicionales derivadas de la compresión renal. En el segundo de los cadáveres, el correspondiente al varón, se observaron cambios morfológicos similares si bien el tamaño exhibido por los riñones era aún mayor, con pesos de 300 y 310 gr. El resto de las vísceras abdominales estaban comprimidas contra el diafragma. En ambos casos se realizó un estudio histológico detallado, centrado especialmente en los riñones en los que se demostraron múltiples quistes de distintos tamaños con morfología sacular a nivel cortical. Dichos quistes ocupaban la mayor parte del parénquima corticomedular si bien las zonas conservadas no mostraban alteraciones significativas salvo inmadurez focal. Dichos quistes estaban tapizados por un epitelio simple que variaba desde plano o cúbico. Los quistes medulares, de menor tamaño y más redondeados estaban tapizados por un epitelio de predominio cúbico. Después de las renales, las alteraciones más llamativas se encontraban en el hígado donde se observaron proliferación y dilatación, incluso quística, de los ductos biliares a nivel de los espacios porta. Con tales hallazgos se emitió en ambos casos el diagnóstico de enfermedad poliquística renal autosómica recesiva infantil.



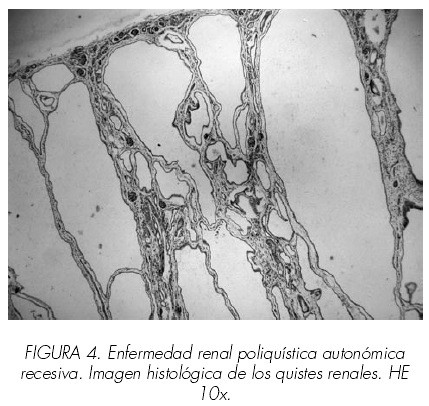


Discusión
La enfermedad renal poliquística infantil se ha dividido tradicionalmente en dos entidades diferenciadas en cuanto a pronóstico y tratamiento: la autosómica recesiva, propia de recién nacidos e infantes y la dominante, que si bien se puede manifestar en las mismas edades, es característica de edades más avanzadas. Las características clínicas, radiológicas y macroscópicas de ambas son similares teniendo que ser necesario en ocasiones recurrir al estudio familiar y genético para consolidar un diagnóstico de certeza, importante de cara al consejo que se le debe dar a las familias.
Desde el punto de vista morfológico, los riñones del recién nacido presentan gran tamaño, son no funcionantes y muestran quistificación difusa. De manera concomitante el hígado presenta en la mayoría de las ocasiones una proliferación de conductillos biliares en las áreas porta del hígado. Mientras las manifestaciones renales son más importantes y relevantes que las hepáticas en el recién nacido, esta relación se va invirtiendo conforme las manifestaciones ocurren a edades más avanzadas de modo que en adultos la única manifestación puede consistir únicamente en fibrosis portal (la llamada fibrosis hepática congénita). En el recién nacido el tamaño renal puede llegar a ser alarmante comprimiendo en la mayoría de los casos los pulmones, que a su vez son hipoplásicos y conducen rápidamente a una situación de distrés respiratorio severo, principal causante de la muerte en estos pacientes. A su vez, la hipo o afunción renal condiciona oligoamnios y en consecuencia compresión facial y facies Potter. Por su parte, aquellos pacientes que logran superar la infancia van a mostrar un predominio de las lesiones hepáticas en forma de fibrosis periportal laxa y proliferación de conductillos biliares, situación que ha sido descrita en la literatura como fibrosis hepática congénita, causa de hipertensión portal y por tanto factor predisponerte al desarrollo de esplenomegalia, principalmente, y de otras manifestaciones clínicas derivadas de la misma tales como varices en el tubo digestivo, ulceración así como las propias del posible fallo hepático (3,6).
La silueta renal suele estar respetada si bien a los cortes seriados se observa un parénquima de aspecto esponjoso debido a la presencia de múltiples estructuras quísticas de pequeño tamaño que al estudio microscópico tienen un aspecto sacular y están tapizados por epitelio cúbico. Dichos quistes se localizan tanto a nivel medular como cortical siendo estos últimos más alargados y grandes que los medulares, más redondeados. El estudio con lectinas identifica los quistes como originados en los túbulos colectores y/o segmentos tubulares distales. La arquitectura renal interquística está respetada aunque se aprecia edema a nivel cortical. En el hígado se aprecia proliferación y menos frecuentemente quistificación de conductos biliares.
Cuanto más tardías sean las manifestaciones de la enfermedad menor será el número de quistes y mayor la atrofia y los cambios intersticiales en el riñón si bien los cambios hepáticos se mantendrán más o menos constantes a excepción del incremento en la fibrosis.
Han sido varios los estudios que han intentado tipifificar la genética de esta entidad logrando identificar el gen PKHD1, situado en el cromosoma 6p21-23 y responsable de la codificación de la fibrocistina la cual parece estar relacionada con la diferenciación de los túbulos colectores y los conducto biliares; de ahí la alteración de ambos con las mutaciones del citado gen (1,2,4,5). Estudios en animales utilizando esta molécula como diana parecen estar obteniendo buenos resultados en frenar la progresión de la enfermedad.
Bibliografía y lecturas recomendadas (*lectura de interés y **lectura fundamental)
*1. SWEENEY, W.E.; AVNER, E.D.: Molecular and cellular pathophysiology of autosomal polycystic kidney disease (ARPKD). Cell Tissue Res.; 326:671, 2006. [ Links ]
2. BERGMANN, C.; FRANK, V.; KUPPER, K. y cols.: Functional analysis of PKHD1 splicing in autosomal recessive polycystic kidney disease. J. Hum. Genet.; 51:788, 2006. [ Links ]
3. BASMAISON, O.; LIUTKUS, A.; MICHEL, L. y cols.: Inherited renal diseases and prenatal diagnosis. Arch Pediat.; 13: 727, 2006. [ Links ]
4. BERGMANN, C.; SENDEREKMJ, SEDLACEK y cols.: Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J. Am. Soc. Nephrol.; 14: 76, 2003. [ Links ]
5. WARD, C.J. y cols.: The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor like protein. Nat. Genet.; 30: 259, 2002. [ Links ]
*6. KUMAR, V.; ABBAS, A. K. y FAUSTO, N.: Robbins and Cotran Pathologic Basis of the Disease; 967. Elsevier, Madrid 2005. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Francisco Javier Torres Gómez
C/ Matahacas, 18ª - 1B
41003 Sevilla. (España).
javiertorresgomez@yahoo.es
Trabajo recibido: 13 de diciembre 2006.

