










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkArchivos Españoles de Urología (Ed. impresa)
versión impresa ISSN 0004-0614
Arch. Esp. Urol. vol.62 no.6 jul./ago. 2009
Angiomiolipoma epitelioide: una variante rara del angiomiolipoma renal
Epithelioid angiomyolipoma: A rare variant of renal angiomyolipoma
Juan Carlos Astigueta1, Milagros A. Abad2, Mariela R. Pow-Sang3, Carlos Morante3, Luís Meza3, Víctor Destefano3 y Richard Dyer2
Servicio de Cirugía Urológica1. Instituto Regional de Enfermedades Neoplásicas Norte. Trujillo. Perú. Departamentos de Patología Oncológica2 y Urología Oncológica3. Instituto Nacional de Enfermedades Neoplásicas. Lima. Perú.
Dirección para correspondencia
RESUMEN
Objetivo: Presentar un caso de angiomiolipoma (AML) variante epitelioide, primario renal, su asociación con Esclerosis Tuberosa (ET) y revisión de la literatura.
Métodos: Presentamos el caso de un paciente varón de 12 años con antecedente de retardo en el desarrollo psicomotor, crisis epilépticas tónico clónicas y estigmas cutáneos, todo esto compatible con ET. Debuta con hematuria macroscópica y dolor abdominal, encontrándose en la tomografía tumor que compromete dos tercios superiores del riñón izquierdo. Fue sometido a nefrectomía radical izquierda. La anatomía patológica, corroborada con estudios de inmunohistoquímica informó la presencia de AML, variante epiteliode.
Se reviso la bibliográfica existente sobre esta variante poco común y su comportamiento maligno.
Resultados: La presencia de la variante epitelial es poco frecuente pero debe tenerse en cuenta por su comportamiento maligno y por lo tanto diferente pronóstico y seguimiento comparado al AML clásico.
Conclusiones: El AML renal es un tumor benigno, poco común, que representa un reto para el diagnóstico clínico e histopatológico. A pesar del gran tamaño que puede alcanzar, la bilateralidad, la multiplicidad de las lesiones y/o el compromiso linfático regional, no se ha demostrado su potencial maligno. Sin embargo, en los últimos años se ha descrito la variante epitelioide, entidad rara de comportamiento agresivo, difícil caracterización histológica y pobre pronóstico.
Palabras clave: Angiomiolipoma epiteliode. Riñón. Esclerosis tuberosa.
SUMMARY
Objective: We present a case of primary renal epithelioid angiomyolipoma, its association with tuberous sclerosis and review the literature.
Methods: We present the case of a 12 year-old male with past medical history of tuberous sclerosis, characterized by developmental delay, tonic and clonic seizures, and cutaneous abnormalities. He presented with macroscopic hematuria and abdominal pain. CT scan of the abdomen showed the presence of a left renal tumor. He underwent left radical nephrectomy. Pathologic study of the specimen showed primary renal epithelioid angiomyolipoma, corroborated by immunohistochemistry staining. Review of the literature was performed for this rare variant and its malignant potential.
Results: The presence of this epithelial variant is rare and must be taken into account because of its malignant potential and, thus, with different prognosis and follow up, compared to classical angiomyolipoma.
Conclusions: Renal angiomyolipoma is an uncommon benign tumor, representing a challenge for clinical and pathological diagnosis. Despite the big size they can reach, as well as bilaterality, multiplicity of lesions and/or lymphatic regional involvement, its malignant potential has not been established. Nevertheless, the epithelioid variant has been described recently, a rare entity with aggressive behavior, difficult histological characterization and poor prognosis.
Key words: Epithelioid angiomyolipoma. Kidney. Tuberous sclerosis.
Introducción
El angiomiolipoma (AML) renal es un tumor poco frecuente, de naturaleza benigna, descrito por primera vez por Morgan en 1951. Está constituido por tres componentes: tejido adiposo maduro, vasos sanguíneos y músculo liso (1,2). Sin embargo se describe una variante histológica agresiva, de comportamiento maligno denominada Angiomiolipoma Epitelioide (AMLE); entidad recientemente separada del resto de AML típicos, asociada en más de la mitad de los casos a Esclerosis Tuberosa (ET), mutaciones en el gen p53 y a una elevada tasa de metástasis a distancia (3,4).
Hasta la fecha son pocos los casos publicados en la literatura (5-7). Nosotros presentamos el primer caso diagnosticado en la institución.
Presentación del caso
Paciente varón de 12 años, con antecedentes de larga data de retardo en el desarrollo psicomotor y crisis epilépticas tónico clónicas generalizadas tratadas con fenitoina y carbamazepina.
Ingresa al Instituto con historia de tres semanas de hematuria macroscópica con coágulos y dolor abdominal predominantemente en hipogastrio. En la tomografía axial computarizada de abdomen y pelvis se observó masa de aspecto heterogéneo de 80 x 64 x 56 mm. que comprometía los dos tercios superiores del riñón izquierdo. Se completaron los estudios imagenológicos con una radiografía de tórax que fue normal y una tomografía cerebral en la que se observó alteraciones hipodensas cortico-subcorticales asociadas a múltiples calcificaciones gruesas periventriculares (Figura 1).
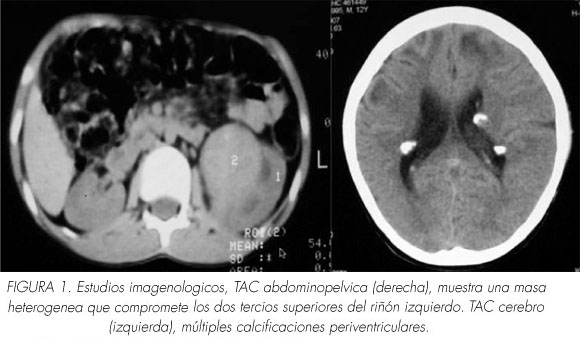
Al examen físico se encuentra en un paciente despierto que no obedecía órdenes, con severa alteración de las funciones cerebrales superiores sin déficit motor. El fondo de ojo no evidenció lesiones retinianas. En el rostro, a nivel de la nariz, pliegues nasogenianos y mentón presentaba lesiones pápulo eritematosas múltiples de hasta 2 mm. A la palpación se encontró en todo el hemiabdomen izquierdo una masa de tamaño no delimitable.
Se planteó el diagnóstico de Angiomiolipoma renal izquierdo asociado a esclerosis tuberosa. Fue sometido a laparotomía exploradora con nefrectomía radical izquierda y linfadenectomía paraaórtica. Se encontró un tumor blando de aproximadamente 10 cm. localizado en la mitad superior del riñón izquierdo, los ganglios regionales no impresionaron estar comprometidos por enfermedad. Evolucionó favorablemente, siendo dado de alta a los 3 días de operado.
La patología informó recibir una pieza operatoria de 250 gr. de 11.0 x 7.4 x 4.2 cm. Al corte el parénquima renal se encontraba reemplazado en el 90% por formación tumoral de 8.0 x 7.0 x 2.3 cm. de color marrón pardo, de aspecto hemorrágico, necrótico que infiltraba y destruía el sistema pielocalicial (Figura 2). El uréter midió 4.6 x 0.8 cm. sin alteraciones macroscópicas significativas. Los bordes quirúrgicos se encontraron libres de neoplasia. Ganglios paraaórticos de aspecto congestivo.

Microscópicamente se observó una lesión tumoral in-filtrativa de bordes irregulares, muy celular constituída fundamentalmente por células poligonales de aspecto
epitelioide, la mayoría con citoplasma claro, entremezcladas con células de citoplasma granular, eosinofílico y otras multinucleadas semejantes a las células ganglionares. Éstas células mostraban atipia nuclear, núcleos vesiculosos, nucleolos prominentes y mitosis (0-5 mitosis / 10 CAP), además de áreas de hemorragia y necrosis. No se observó invasión vascular ni perineural. El uréter, la grasa perirenal y los bordes quirúrgicos se encontraron libres de neoplasia. Con esta histomorfología se planteó el diagnóstico de angiomiolipoma epitelioide, el mismo que se confirmó con pruebas de inmunohistoquímica, cuyo resultado se presenta en la Tabla I.

Discusión
El AML renal, también conocido como hamartoma, es un tumor benigno relativamente infrecuente que aparece en el 0.3% de la población general y representa el 3% de las masas renales sólidas. Pertenece a la familia de lesiones caracterizadas por la proliferación de células epitelioides perivasculares (PEC) y está constituido, en proporción variable por tres estirpes celulares: tejido adiposo maduro, músculo liso y vasos sanguíneos irregulares (1,2). Habitualmente se presenta bajo dos formas clínicas: como un tumor único grande en mujeres de mediana edad, o bien asociado a la Enfermedad de Bourneville o Esclerosis tuberosa donde suele ser bilateral, múltiple y con mayor incidencia en varones jóvenes, representando entre el 15 y 20% de los casos en las diversas series (3-5).
La Esclerosis Tuberosa (ET) es una enfermedad hereditaria cuyo diagnóstico se puede realizar en distintas etapas de la vida. En el lactante se inicia con espasmos de flexión, hipsiarritmia y manchas acrómicas en la piel. En niños mayores y adolescentes, como nuestro paciente, se presenta con epilepsia, angiofibromas faciales y presencia de calcificaciones intracraneales en la región de las paredes ventriculares (6,7). También compromete órganos como riñón, hígado, cerebro, corazón, ojos, pulmones y hueso. Las manifestaciones renales más frecuentes son los angiomiolipomas, quistes, carcinoma renal o la combinación de estas lesiones (811). La asociación AML - carcinoma de células renales es un hallazgo inusual, hasta el 2001 se conocían en la literatura aproximadamente 50 reportes de casos. Dada la escasez de casos publicados, todavía no es posible saber en que medida influye en el pronóstico esta asociación (12).
Los AML están presentes entre el 50 al 80% de los casos de ET y su asociación es más estrecha con la variante epitelioide de AML renal, una entidad de reciente descripción, con un poco más de doce casos reportados en la literatura y que se caracteriza por su agresividad. Se sabe que más de la mitad de pacientes con AMLE tienen ET (2,6).
El AMLE afecta igualmente ambos sexos, con un promedio de edad al momento del diagnóstico de 38 años. Los pacientes son sintomáticos, por lo general presentan dolor en flanco, masa palpable y menos del 15% puede debutar con insuficiencia renal debido a la comprensión y reemplazo del parénquima renal. Es un tumor que trae problemas diagnósticos siendo muchas veces interpretado como un carcinoma de células renales o un sarcoma de alto grado. Los estudios de imágenes simulan carcinoma de células claras por la escasez del tejido adiposo (7,8).
Macroscópicamente son tumores de mediano a gran tamaño, de color amarillento anaranjado con extensas áreas de hemorragia y necrosis. Puede haber extensión extrarenal o compromiso de la vena cava o vena renal. Microscópicamente es una lesión de carácter infiltrativo, muy celular, constituida fundamentalmente por células poligonales, con citoplasma vacuolado, eosinofílico granular o claro, con abundante glucógeno, que Apitz describió como epitelioides y otras multinucleadas semejantes a las células ganglionares. También puede encontrarse una pequeña proporción de células fusocelulares. Las células muestran anaplasia nuclear, actividad mitótica variable con presencia de figuras atípicas, invasión vascular, necrosis y respuesta inflamatoria. La hemorragia es prominente. Unos pocos casos tienen áreas focales de AML clásico. La mayoría, al igual que en el caso que presentamos, carecen de los elementos típicos del AML (9).

La inmunohistoquímica es de vital importancia para caracterizar este tumor. La presencia de inmunomarcación positiva para HMB45, HMB50, CD117, CD63 y la negatividad para marcadores epiteliales y citoqueratinas confirman el diagnóstico (2). También se ha descrito una expresión variable para marcadores musculares lisos (actina músculo liso y actina músculo específico). Genéticamente comparten la misma alteración que el AML clásico: la pérdida alélica del brazo corto del cromosoma 16p (TS2), pero sólo se han detectado mutaciones en el gen p53 en la variante epitelioide, lo que sugiere un rol importante en su comportamiento maligno. En aproximadamente un tercio de casos de AMLE se han descrito metástasis a ganglios linfáticos, hígado, pulmón, hueso y médula, pero hasta la fecha no se ha identificado ningún parámetro patológico que se correlacione directamente con sobrevida. Sin embargo, los tumores con necrosis, actividad mitótica y anaplasia nuclear tienen un comportamiento más agresivo (1,2).
El manejo de los angiomiolipomas está ampliamente discutido en la literatura (9,13-15). La pauta terapeútica más aceptada es el algoritmo de Oesterling et al. basado en la presentación clínica, el tamaño y la bilateralidad del tumor. De esta forma en tumores asintomá-ticos se realizarán controles periódicos con ecografía y/o TAC cada seis o doce meses según el tamaño sea mayor o menor de 4 cm., respectivamente. En los tumores sintomáticos y/o bilaterales se intentará una embolización arterial selectiva o cirugía renal conservadora. La nefrectomía radical quedará reservada a aquellos casos con inestabilidad hemodinámica por hemorragia masiva, tumores de gran tamaño o coexistencia de carcinoma en el mismo riñón; criterios que se consideraron en el manejo terapéutico de este caso (14,15).
Conclusión
El Angiomiolipoma (AML) renal es un tumor benigno, poco común, que representa un reto para el diagnóstico clínico e histopatológico. A pesar del gran tamaño que puede alcanzar, la bilateralidad, la multiplicidad de las lesiones y/o el compromiso linfático regional, no se ha demostrado su potencial maligno. Sin embargo, existe la variante Epitelioide, entidad poco común, de comportamiento agresivo, de difícil caracterización histológica y pobre pronóstico. Dicha variante debe tenerse en cuenta en el diagnóstico y tratamiento considerando que la evolución y seguimiento son muy diferentes a la del AML clásico.
Bibliografía y lecturas recomendadas (*lectura de interés y **lectura fundamental)
*1. Martigoni G, Amin MB. Angiomyolipoma. En Tumours of the urinary system and male genital organs. Lyon France. WHO IARC; 2004: 63-67. [ Links ]
*2. Amin MB. Epithelioid angiomyolipoma. En Tumours of the urinary system and male genital organs. Lyon France. WHO IARC; 2004: 68-69. [ Links ]
3. Eble JN, Amin MB and Young RH. Epithelioid angiomyolipoma of the kidney: a report of five cases with a prominent and diagnostically confusing epithelioid smooth muscle component. Am J Surg Pathol. 1997; 21: 1123-1130. [ Links ]
**4. Serrano Frago P, Del Agua Arias Camison C, Gil Sanz MJ, Allue Lopez M, Gonzalvo Ibarra A, Plaza Mas L et al: Controversies related to epithelioid variant of renal angiomyolipoma: a review of the literature. Urology 2006; 67: 846.e3-5. [ Links ]
5. Acikalin MF, Tel N and Oner U. Epithelioid angiomyolipoma of the kidney. Int. J Urol. 2005; 12(2): 204-207. [ Links ]
6. Bjornsson J, Short MP, Kwiattkowski DJ and Hens-ke EP. Tuberous sclerosis associated renal cell carcinoma, clinical, pathological and genetic features. Am J Surg Pathol. 1996; 149(4): 1201-1208. [ Links ]
7. Roach ES, Gomez MR and Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. JChild Neurol 1998; 13: 624-628. [ Links ]
*8. Nelson CP and Sanda MG. Contemporary diagnosis and management of renal angiomyolipoma. J Urol 2002; 168: 1315-1325. [ Links ]
**9. Lane BR, Aydin H, Danforth T, Zhou M, Remer E, Novick AC and Campbell SC: Clinical correlates of renal angiomyolipoma subtypes in 209 patients classic, fat poor, tuberous sclerosis associated and ephitelioid. J Urol 2008; 180: 836-843. [ Links ]
10. Christiano AP, Yang X and Gerber GS: Malignant transformation of renal angiomyolipoma. J Urol 1999; 161: 1900-1901. [ Links ]
11. Kawaguchi K, Oda Y, Nakanishi K, Saito T, Tamiya S, Nakahara K et al: Malignant transformation of renal angiomyolipoma: a case report. Am J Surg Pathol 2002; 26: 523-529. [ Links ]
12. Jiménez RE, Eble JN, Reuter VE, Epstein JI, Folpe AL, de Peralta-Venturina M et al: Concurrent angiomyolipoma and renal cell neoplasia: a study of 36 cases. Mod Pathol 2001; 14: 157-163. [ Links ]
*13. Danforth TL, Lane BR and Novick AC: Conservative management of giant symptomatic angiomyolipoma in patients with tuberous sclerosis complex. BJU Int 2007; 100(4): 794-797. [ Links ]
*14. Oesterling JE, Fishman EK, Goldman SM and Marshall FF: The management of renal angiomyolipoma. J Urol 1986; 135: 1121-1124. [ Links ]
15. Dickinson M, Ruckle H, Beaghler M and Hadley HR: Renal angiomyolipoma: optimal treatment based on size and symptoms. Clin Nephrol 1998; 49(5): 281-286. [ Links ]
 Dirección para correspondencia:
Dirección para correspondencia:
Milagros Abad Licham, MD.
Departamento de Patología Oncológica
Instituto Nacional de Enfermedades Neoplásicas
Av. Angamos Este 2520
Surquillo, Lima. (Perú).
e-mail: milagros.abad@hotmail.com,
jca_astigueta@hotmail.com
Recibido: 23 de diciembre 2008.

