










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkArchivos Españoles de Urología (Ed. impresa)
Print version ISSN 0004-0614
Arch. Esp. Urol. vol.63 n.1 Jan./Feb. 2010
Biología molecular en el cáncer de próstata
Molecular biology in prostate cancer
Marco Antonio Arap
Hospital de Clínicas. Universidad de São Paulo.
Núcleo Avançado de Urología. Hospital Sírio Libanês. São Paulo. Brasil.
Dirección para correspondencia
RESUMEN
El cáncer de próstata se ha convertido en uno de los tumores más frecuentemente diagnosticado en los hombres y es una de las principales causas de muerte en hombres mayores de 50 años. Debido al desarrollo de técnicas de biología molecular utilizadas en estudios de cáncer de próstata recientes, se han descubierto muchos aspectos nuevos de la enfermedad que pueden ayudar en el diagnóstico, tratamiento e incluso para establecer el pronóstico de estos pacientes. Por lo tanto, y, a pesar de que aún no es común en la práctica clínica, hay varias técnicas de biología molecular que, con frecuencia deberían discutirse con otros médicos e incluso con el paciente. En este artículo revisamos algunos de los instrumentos más importantes utilizados en biología molecular y sus descubrimientos recientes en el estudio del cáncer de próstata.
Palabras clave: Cáncer de próstata. Biología molecular. Aplicaciones.
SUMMARY
Prostate cancer has become one of the most frequently diagnosed tumors in men and is one of the leading causes of death in men over 50 years old. Due to the development of molecular biology techniques used in recent prostate cancer studies, many new aspects of the disease are being discovered and may help in diagnosis, treatment and even to establish prognosis for these patients. Therefore, despite not yet common in clinical practice, several molecular biology techniques frequently need to be discussed with other physicians and even with the patient. In this article, we review some of the most important tools used in molecular biology and their recent discoveries in the study of prostate cancer.
Key words: Prostate cancer. Molecular biology. Applications.
Introducción
El cáncer de próstata (CP) es uno de los principales problemas de salud pública en todo el mundo. En los Estados Unidos se espera que serán diagnosticados 190.000 nuevos casos y más de 27.000 hombres morirán de la enfermedad en 2009 (1). A pesar de la utilización actual del antígeno prostático específico (PSA) y del tacto rectal, dos estudios recientes informan sólo una ligera reducción de la mortalidad del CP (2) o ninguna reducción (3) relacionadas con esa práctica. Por otra parte, todavía no conocemos todos los factores que influyen en el inicio del cáncer de próstata, así como por qué algunos tumores de los pacientes progresarán de forma latente a enfermedad invasiva.
En cuanto a otros tipos de tumores, se han descrito muchas lesiones pre-malignas en el CP. Neoplasias intraepitelial prostática de alto grado (PIN) (4, 5) y la atrofia inflamatoria proliferativa (PIA) (4) son potencialmente precursoras del cáncer de próstata. Es evidente, por tanto, que muchas otras características de la enfermedad, tales como marcadores moleculares y genes implicados en la progresión, todavía están por descubrir, a fin de aclarar los mecanismos de inicio del CP, progresión y, eventualmente, reducir la mortalidad asociada. La biología molecular ofrece muchas herramientas que pueden ayudar a una mejor comprensión del CP. Este artículo revisa algunos de los campos más importantes de la investigación de biología molecular en el CP y sus descubrimientos más recientes.
Perfil de expresión genético
La investigación sobre los orígenes genéticos del PC se ha incrementado de forma exponencial, debido principalmente a los nuevos microarrays genéticos de alto rendimiento y tecnologías de secuenciación. Después de la introducción de microarrays de expresión genética, estudios con muestras de tumores y líneas celulares se evaluaron aspectos moleculares del cáncer de próstata, con el fin de poner de relieve las características genéticas de la enfermedad (6-8). Estos estudios identificaron genes que participan en las principales vías de regulación (9), como la réplica y reparación del ADN e incluso establecieron la correlación con los resultados clínicos (10).
A fin de validar los genes candidatos, los científicos utilizaron PCR cuantitativo en tiempo real, ADN arrays, inmunohistoquímica, y microarrays de tejidos. En consecuencia, se han publicado muchos estudios importantes de perfil de expresión, algunos muestran relación entre el cáncer de próstata y factores de crecimiento (11), proteínas chaperonas y marcadores tisulares que se utilizan actualmente para la evaluación de las biopsias de próstata (8, 12). Además, se ha utilizado la inmunohistoquímica para establecer la regulación de genes en un intento de identificar los biomarcadores del CP. A pesar de todos los esfuerzos, hasta la fecha no existen marcadores moleculares o histológicos utilizados de forma rutinaria que predigan el curso de la enfermedad, quizás debido a otras características de los pacientes y a la importancia de la heterogeneidad del CP a la hora de definir el pronóstico. Una posible solución utilizada para superar la heterogeneidad del CP es la captura por microdisección láser y los procedimientos de amplificación adicional. La captura por microdisección láser es una técnica que permite el aislamiento y la recuperación de poblaciones de células diferentes en un solo tejido, ya sea benigno o maligno. De hecho, incluso se puede diseccionar, aislar y recuperar una sola célula, utilizando la precisión del láser, una mejora importante respecto a la microdisección estándar. Después de la microdisección de tejidos, se pueden amplificar para su posterior estudio cantidades muy pequeñas de ARN.
Las firmas de expresión genética se han descrito por primera vez en 2006, para la estadificación patológica del PC como una correlación molecular con el sistema de Gleason. Utilizando la microdisección de tejidos se obtuvieron y combinaron con las células prostáticas benignas, grupos específicos de células cancerosas correspondientes a los patrones de Gleason más frecuentes (3, 4 y 5), procedentes de 29 muestras de prostatectomía radical. Se identificó un panel de 86-genes y capaz de distinguir carcinomas de grado bajo (Gleason 3) y de alto (Gleason 4 y 5) (13). Además, este modelo tenía un 76% de precisión cuando se validó con un grupo independiente de carcinomas de próstata. En otro estudio, el gen DD3 fue descrito como altamente expresado en la microdisección de cáncer de próstata (14). El gen DD3 fue posteriormente llamado antígeno de cáncer de próstata 3 (PCA3) y, en un estudio diseñado para evaluar su potencial de diagnóstico en más de 100 hombres, se detectó PC3 en hasta un 95% de las muestras de CP y su expresión resultó ser más de 60 veces mayor en los tejidos prostáticos malignos que en los benignos (15). A nivel celular, la determinación del PCA3 puede separar las células prostáticas benignas de las malignas con una precisión del 100%. Actualmente se está investigando a fondo la sobreexpresión de este gen en los fluidos corporales que contienen material celular prostático (como el semen y orina), con los primeros estudios que muestran niveles superiores de PCA3 en orina al PSA en el diagnóstico del CP (16-19).
Mutaciones en el cáncer de próstata
Se cree que es crucial la inestabilidad genómica en la carcinogénesis de la próstata, así como en otros cánceres. La acumulación de los polimorfismos genéticos que regulan la proliferación y la muerte celular se han observado durante muchos años en el CP. La heterogeneidad de la expresión génica en el CP es muy común y debida a muchas aberraciones cromosómicas diferentes. En las últimas décadas, estas anomalías han sido estudiadas con muchas técnicas, como la pérdida de heterocigosidad (LOH) y perfiles de ADN. Las innovaciones en la citogenética molecular, tales como la hibridación genómica comparada (CGH) y la hibridación in situ con fluorescencia (FISH) han mostrado algunas de las predominantes regiones cromosómicas implicadas en la carcinogénesis del CP. Las alteraciones más comunes son las pérdidas en 1p, 6q, 8p, 10q, 13q, 16q, y 18q y las ganancias en 1q, 2p, 7, 8q, 18q y Xq. Se ha sido utilizado FISH para descubrir los genes objetivos de algunas de estas alteraciones, que podrían utilizarse como marcadores moleculares de la enfermedad, como la AR (Xq12), EIF3S3 (8q23) y MYC (8q24) (20).
Más recientemente, un estudio diseñado para comparar la precisión del array comparativo de hibridación genómica (aCGH) con el CGH convencional y el análisis LOH se ha demostrado que aCGH es un instrumento poderoso y preciso para la detección completa de las pérdidas en las secuencias de ADN (21). La ventaja de aCGH es la capacidad para probar un mayor número de muestras con el fin de identificar las características genéticas comunes, así como una mayor precisión que el CGH convencional (21). En otro estudio, una evaluación aCGH de líneas de células prostáticas, xenoinjertos de cáncer de próstata y adenocarcinomas identificados primarios y metastásicos, identificó 3 genes sobreexpresados en el cáncer de manera significativa en comparación con el tejido prostático normal, que puede considerarse marcadores putativos de progresión para CP (PDP, situado en 8q22.1, PABPC1 situado en 8q22.3, y KIAA0196 ubicado en 8q24.13) (22).
Otro estudio aCGH muy interesante, compuesto por 64 pacientes previamente sometidos a prostatectomía radical, que estaban en riesgo intermedio o alto de recurrencia post-operatoria se identificaron 40 marcadores candidatos asociados con potencial metástasis. En concreto, la pérdida en 8p23.2 estaba relacionada con la enfermedad en etapa avanzada y el aumento en 11q13.1 resultó ser un factor predictivo de recidiva postoperatoria, independientemente de su grado y estadio (23). El mismo grupo comparó clones identificados por el aCGH con el nomograma de Kattan, en relación con los resultados bioquímicos. Este estudio preliminar reveló una exactitud de 78% de los clones para detectar firmas genómicas de metástasis en los tumores primarios, en comparación con una exactitud de 75% del nomograma de Kattan (24). Por lo tanto, se deben diseñar otras cohortes de validación más numerosas con el fin de evaluar estas alteraciones genéticas como marcadores de resultados para el CP.
Las mutaciones somáticas acumuladas durante la carcinogénesis pueden ser usadas para los tratamientos farmacológicos específicos, ya que muchas proteínas expresadas en las células del cáncer son diferentes a las de correlación normal. Esto ya está siendo estudiado en la próstata y en otros tipos de cáncer (25). El gen KLF6 (Krüppel-como factor de transcripción del dedo de zinc) es un gen supresor de tumores que con frecuencia se inactiva por la pérdida de heterocigosidad (LOH), la mutación somática y/o disminución de la expresión en el cáncer. Codifica una familia de proteínas generadas a través de splicing alternativo que participa en la regulación del desarrollo y progresión del cáncer. El empalme alternativo del gen KLF6 tiene como resultado la producción de al menos cuatro isoformas de corte y empalme alternativo. La variante de empalme KLF6 1 (KLF6-SV1) es una variante oncogénica sobreexpresada en la próstata y otros cánceres, que ha demostrado ser biológicamente activa y que promueve el crecimiento y difusión del tumor. En un estudio multi-institucional de 3411 hombres, una línea germinal KLF6 polimorfismo de un solo nucleótido se asoció con un aumento del riesgo relativo de cáncer de próstata en los hombres (26). La KLF6-SV1 también conduce a la disminución de la expresión de p21 y a aumentar el crecimiento celular, así como una regulación ascendente en el tumor en comparación con el tejido prostático normal. Además, otros estudios demostraron que KLF6 induce la apoptosis en las células de CP (27), y la inhibición del KLF6-SV1 da como resultado la regresión del tumor "in vivo" (28). En conjunto, estos datos sugieren que la familia KLF6 puede ser utilizada para el tratamiento específico de cáncer de próstata.
Utilizando un enfoque bioinformático (atípico perfil de análisis de cáncer - COPA), Tomlins et al., identificaron los genes que están sobreexpresados en un subgrupo de tumores de próstata (29). Utilizando el COPA, se encontró que el 5 'UTR del gen TMPRSS2 andrógeno-regulado se fusionó con genes de la familia ETS de transcripción, dando lugar a la sobreexpresión de los factores de transcripción on-cogénico. También demostraron en una línea celular de cáncer de próstata que la expresión de ERG está regulada por los andrógenos. Por otra parte, un estudio reciente mostró una tendencia significativa en la frecuencia de las fusiones TMPRSS2-ERG en diversos tejidos: 2,4% en la hiperplasia benigna de próstata, 20% en neoplasia de alto grado intraepitelial prostá-tica y el 50% en CP localizado (30). Otros autores también han confirmado la presencia de las fusiones recurrentes de genes (31-34), apoyando su papel potencial como marcadores precoces del CP.
Epigenética
Muchas alteraciones epigenéticas contribuyen a la formación del cáncer de próstata, como con otros cánceres humanos. Tal vez una de las características más interesantes de los cambios epigenéticos es la reversibilidad, ya que la secuencia de ADN se mantiene intacta. Cabe destacar que los cambios epigenéticos generalmente aparecen antes y más consistentemente durante la carcinogénesis. La hipermetilación del ADN es la anomalía epigenética más común del cáncer. Se piensa generalmente que la carcinogénesis puede estar influenciada por el silenciamiento de genes supresores de tumores, especialmente debido a la hipermetilación de islas CpG en sus regiones promotoras (35). En consecuencia, la hipermetilación del ADN puede ser utilizado como un objetivo para la clonación de nuevos genes supresores de tumor.
La hipermetilación es responsable de la inactivación de muchos genes en el cáncer de próstata, como el APC (36), CDH1 (37), MDR1 (38) y RASSF1A (39). Sin embargo, sólo unos pocos genes son candidatos potenciales, como marcadores tumorales para el diagnóstico precoz y la evaluación de riesgo de CP. Chung et cols., utilizando ampliación de isla CpG metilada, junto con el análisis representativo de la diferencia, de 8 genes seleccionados (NKX2-5; SPOCK2 r; GALR2; LSTN1; NSE1; DPYS; FOXN4; SLC16A12) con más metilación en cáncer de próstata en comparación con la próstata normal adyacente. La combinación de algunos de estos genes fue capaz de diferenciar cáncer de la próstata normal hasta con un 96% de precisión, ofreciendo nuevos posibles biomarcadores para la detección del CP (40).
Ellinger et al., también estudiaron la hipermetilación en el PC utilizando una metilación-específica por reacción en cadena de la polimerasa (41). Se evaluaron controles de nueve localizaciones genéticas en 80 pacientes con CP y 26 con hiperplasia benigna de la próstata (HPB). La hipermetilación fue más frecuente en el CP que en las muestras de HPB. La hipermetilación en una única localización genética no se correlacionó con ninguna variable clínico-patológicas. Por otra parte, la hipermetilación en dos genes se correlacionó significativamente con el estadio patológico y/o escala de Gleason. La hipermetilación de TIG1 y GSTP1 fue capaz de distinguir el CP de la HBP con 85% especificidad y 93% de sensibilidad. Además, la hipermetilación del ADN en más de cinco genes, se correlaciona significativamente con la tasa de recurrencia de PSA tras la prostatectomía radical (41).
Muchos otros genes candidatos también se han asociado con la susceptibilidad del CP (HPC1 (42), HPC2 (43), PCAP (44)) y progresión (Hepsin (45), GST-pi (46), p27 (47, 48), E-cadherina (4951), NKX3.1 (52)), utilizando diferentes técnicas de biología molecular. Creemos que en un futuro próximo, se utilizará de forma habitual un perfil molecular de los tumores de próstata para el pronóstico y tal vez para ayudar en la orientación del tratamiento.
Fagos
Los bacteriófagos (o simplemente fagos) son virus de ADN de cadena simple que infectan a las bacterias gram negativas. Las partículas más comunes del fago utilizadas para la investigación es la cadena Fd, que consiste en una cápside cilíndrica de proteína que encierra un genoma ADN de cadena simple con 11 genes y alrededor de 6400 nucleótidos. La partícula de virus está formado por proteínas pVIII (cuerpo viral) y proteínas PIII, PVI, PVII y pXIX (viral final) (53). En la mayoría de los casos, la proteína PIII proteína se utiliza para exponer péptido.
La tecnología de fagos fue originalmente introducida para rastrear los puntos de unión de las inmunoglobulinas (54, 55). La tecnología se basa en un enfoque combinatorio que permite la presentación de colecciones de péptidos en la superficie de fagos filamentosos, que conducirá a la selección de proteínas, incluyendo anticuerpos, con una alta afinidad y especificidad a casi cualquier objetivo, sin nociones preexistentes sobre la naturaleza de los objetivos (56). Hasta 1010 variantes de péptidos exógenos pueden ser introducidos en el genoma del fago y expresados por las proteínas del fago. Además, las partículas del fago puede resistir muchas condiciones difíciles, tales como el pH bajo y bajas temperaturas, sin perder infectividad. De hecho, la mayoría de los protocolos usan pH bajo para disociar fago de un objetivo.
La investigación de los bacteriófagos ofrece la posibilidad de distinguir la especificidad de unión de péptidos de diferentes objetivos, tales como proteínas, tejidos, órganos o incluso animales vivos. Normalmente la selección de afinidad de los péptidos de una biblioteca de fagos (llamada "ciclo de selección") se define en 5 pasos fundamentales: la creación de una biblioteca principal o ampliación de una biblioteca existente, la exposición del fago a un objetivo específico, la eliminación de aglutinantes no específicos (lavado/perfusión), recuperación del fago dirigido a un objetivo por elución o infección bacteriana directa y la amplificación de los fagos recuperados. Esta panorámica se repite, por lo general, varias veces hasta que se selecciona una población de mejores aglutinantes. Al secuenciar el genoma codificando el péptido mostrado, es posible determinar y reproducir su secuencia como péptido recombinante o sintético. De esta manera uno puede determinar finalmente los ligandos específicos y selectivos a los receptores de destino (57).
Durante los últimos años se ha estudiado la selección de los receptores-ligandos en sistemas biológicos complejos, tales como células vivas y animales. Uno de los objetivos más importantes de la próstata que se identificó usando ciclos de selección de los cultivos de células es la proteína-78(GRP78) glucoso-regulada (58). La GRP78 es una proteína chaperona que se inicialmente resultó expresada en el retículo endoplásmico de varios tipos de células (59-61), y que más tarde se demostró que estaba presente en la membrana celular, y que es responsable de la presentación antigénica (62). La GRP78 de inducción está considerablemente aumentada en una variedad de condiciones de tensión celular, tales como glucosa y falta de oxígeno (63). Refleja una respuesta protectora contra la tensión (64, 65) que podría evitar la apoptosis (66). La sobre-expresión GRP78 en los cánceres humanos se puede explicar por el ambiente relativamente hipóxico que se encuentra en los tumores sólidos. Esta sobre-expresión GRP78 desencadena una respuesta inmune contra la proteína que había resultado estar relacionada con el cáncer de próstata andrógeno-independiente, y también a una menor supervivencia en general de los pacientes (58).
Por lo tanto, aparte de la presencia de anticuerpos contra la GRP78 al utilizarse como marcador serológico de cáncer de próstata, la propia proteína podría utilizarse como un objetivo molecular para el tratamiento de la enfermedad. Para evaluar esta hipótesis, se evaluó las interacciones de la proteína-proteína basada en GRP78 en pocillos de microtitulación, las líneas de células del cáncer de próstata, y xenoinjertos y modelos de tumor isogénico en ratones, así como muestras humanas de cáncer de próstata (67). Demostramos que los fagos seleccionados vinculados específicamente a recombinante GRP78 en pocillos de microtitulación, que el origen del fago a xenoinjertos de cáncer de próstata humanos en vivo a través de la administración sistémica y también que reconoce las metástasis del cáncer de próstata humano en el hueso, todo específicamente a través de GRP78 (67).
A continuación, hemos tratado de evaluar si los péptidos sintéticos podrían tener un efecto antitumoral "in vivo". Usando la síntesis de péptidos de Merrifield, Los vínculos de péptidos-GRP78 fueron quimerizados con el D (klaklak)2 motivo proapoptótico (68, 69) y dado a ratones desnudos que tenían xenoinjertos de DU145 derivados de cáncer de próstata y a ratones Balb/c inmunocompetentes de que tenían tumores isogénicos derivados de mama (70). Las dosis semanales por vía intravenosa de péptidos específicos, o bien los controles fueron administrados y los volúmenes tumorales fueron evaluados periódicamente. Los volúmenes tumorales post tratamiento fueron significativamente menores en los ratones tratados con los péptidos específicos cuando se compararon con los controles tratados con los péptidos revueltos y con el vehículo solo. El efecto antitumoral de las quimeras, fue igual en ambos modelos, xenoinjertos de tumores de próstata y de mama isogénicos (67). Estos datos son importantes y relevantes ya que muestran que los efectos no dependen del tipo de tumor (de mama y próstata), origen (humano y del ratón) o estado inmune del huésped.
Juntos, estos estudios han demostrado que el descubrimiento de los sistemas receptor-ligando es la clave para el desarrollo de terapias dirigidas. Además, muestran que la GRP78 es un blanco molecular del tumor que puede ser utilizado para el tratamiento del cáncer de próstata metastásico y cáncer de mama, ambas enfermedades incurables. En consonancia con esta investigación, otros grupos también han mostrado interés en las proteínas de respuesta a la tensión en la próstata (71) y otros cánceres urológicos (71), así como en las enfermedades no malignas (72).
Discusión
Con los años, los métodos más refinados de estudio de los cánceres humanos han contribuido a una notable mejora en la comprensión no sólo la iniciación del tumor, sino también de su progresión y heterogeneidad. Debido a los avances de biología molecular, los científicos entienden ahora que probablemente todos los cánceres se desarrollan a partir de múltiples causas factoriales.
En circunstancias normales, hay un equilibrio en el reemplazo de células dentro de un órgano o tejido. Las nuevas células se generan sólo para reemplazar las antiguas que se pierden debido a cualquier tipo de estrés, como lesiones o hipoxia. La división celular, está muy regulada por las proteínas codificadas por los genes que controlan el crecimiento. En el caso de las aberraciones de ADN, las células pueden perder su equilibrio regulador, que es responsable de la división celular sólo después de recibir una señal adecuada. En el cáncer, los proto-oncogenes y genes supresores de tumores están generalmente mutados, lo que lleva a un crecimiento celular incontrolado. Sin embargo, las células de cáncer, probablemente necesitan tener varias características con el fin de eludir los mecanismos de defensa del huésped y promover cáncer clínico: autosuficiencia en las señales de crecimiento y/o insensibilidad a las señales anticrecimiento (por ejemplo a través de la proteína del retinoblastoma), la evasión de la apoptosis (por lo general debido a la proteína p53 mutada), auto-sostenido angiogénesis autosostenida(como en el factor de regulación del crecimiento vascular endotelial), la invasión de tejidos y el potencial metastásico (por ejemplo a través expresión anormal de E-cadherina).
Como se ilustra en esta revisión, se han descrito los marcadores de candidatos diferentes para la carcinogénesis prostática (Tabla I). Aunque el concepto original de un oncogén simple o gen supresor de tumores ha cambiado debido a la descripción de nuevas mutaciones somáticas y las variantes de empalme alternativo que se traducen en diferencias significativas en función de las proteínas, varias vías fueron aclaradas mediante las clásicas y las nuevas técnicas de biología molecular en los últimos años. Otras técnicas moleculares emergentes como la bio-informática y la proteómica se están aplicando a las muestras de suero, con el fin de identificar los perfiles de proteínas (73). De hecho, hay diferentes enfoques de la proteómica que se utilizan para un perfil de CP, incluyendo la electroforesis bidimensional y la proteómica SEDI-TOF. El primer enfoque utiliza el tamaño y la carga eléctrica de la proteína para separar grandes cantidades de proteína, mientras que la SEDI-TOF permite la caracterización de muestras muy pequeñas, como las obtenidas con microdisección láser. Ambos métodos obtendrán perfiles de proteínas que requeriran identificación adicional.
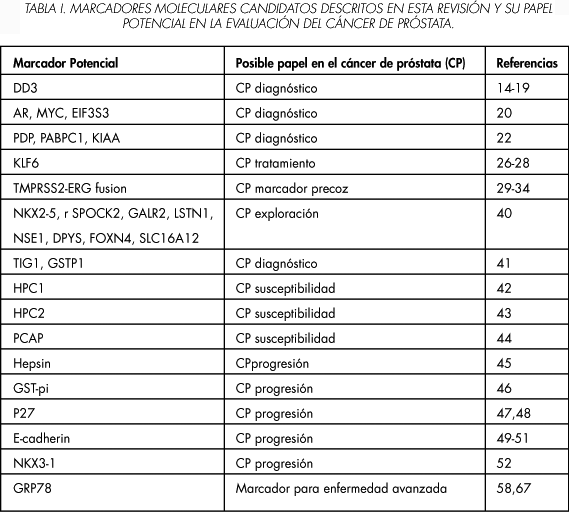
Todas las técnicas y los marcadores mencionados anteriormente representan importantes avances. Tal vez la cuestión más difícil es la traducción entre el trabajo de laboratorio y la comercialización final de tales marcadores. Hasta la fecha, sólo unos pocos está disponibles, como una prueba de orina molecular para el CP3. Por lo tanto, teniendo en cuenta todos los mecanismos que en última instancia conducen a diferentes patrones de expresión génica, es evidente que los urólogos, oncólogos y otros médicos también necesitarán poseer un conocimiento detallado de técnicas moleculares para el desarrollo de ensayos clínicos.
Conclusiones
El cáncer de próstata es una de las patologías más importantes en oncología. No existe una terapia ideal para cualquiera de sus etapas y, aún hoy, muchos pacientes sufren por la propia enfermedad o por los efectos secundarios del tratamiento. La biología molecular abarca diferentes tipos de investigación, tales como la genómica, la proteómica, la epigenética y de fagos, que puede mostrar en el futuro cercano los detalles específicos de la iniciación y progresión de la enfermedad. Los científicos están buscando mejores maneras de diagnosticar el CP, para predecir qué pacientes tendrán recurrencia después del tratamiento inicial, y a establecer mejores marcadores del inicio, progresión y pronóstico de la enfermedad. En este artículo se examinan brevemente algunas de las técnicas de biología molecular implicadas en la investigación del CP.
Bibliografía y lecturas recomendadas (*lectura de interés y **lectura fundamental)
1. Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer Statistics, 2009. CA Cancer J Clin. 2009 27. [ Links ]
**2. Schroder FH, Hugosson J, Roobol MJ, Tammela TL, Ciatto S, Nelen V, et al. Screening and prostate-cancer mortality in a randomized European study. N Engl J Med. 2009 26;360(13):1320-8. [ Links ]
**3. Andriole GL, Crawford ED, Grubb RL, 3rd, Buys SS, Chia D, Church TR, et al. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med. 2009 26;360(13):1310-9. [ Links ]
4. De Marzo AM, Marchi VL, Epstein JI, Nelson WG. Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. Am J Pathol. 1999;155(6):1985-92. [ Links ]
5. Putzi MJ, De Marzo AM. Morphologic transitions between proliferative inflammatory atrophy and high-grade prostatic intraepithelial neoplasia. Urology. 2000 1;56(5):828-32. [ Links ]
6. Chaib H, Cockrell EK, Rubin MA, Macoska JA. Profiling and verification of gene expression patterns in normal and malignant human prostate tissues by cDNA microarray analysis. Neoplasia. 2001;3(1):43-52. [ Links ]
7. Chakrabarti R, Robles LD, Gibson J, Muroski M. Profiling of differential expression of messenger RNA in normal, benign, and metastatic prostate cell lines. Cancer Genet Cytogenet. 2002;139(2):115-25. [ Links ]
8. Xu J, Stolk JA, Zhang X, Silva SJ, Houghton RL, Matsumura M, et al. Identification of differentially expressed genes in human prostate cancer using subtraction and microarray. Cancer Res. 2000 15;60(6):1677-82. [ Links ]
9. LaTulippe E, Satagopan J, Smith A, Scher H, Scardino P, Reuter V, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62(15):4499-506. [ Links ]
*10. Singh D, Febbo PG, Ross K, Jackson DG, Manola J, Ladd C, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer Cell. 2002;1(2):203-9. [ Links ]
11. Bubendorf L, Kolmer M, Kononen J, Koivisto P, Mousses S, Chen Y, et al. Hormone therapy failure in human prostate cancer: analysis by complementary DNA and tissue microarrays. J Natl Cancer Inst. 1999 20;91(20):1758-64. [ Links ]
12. Rubin MA, Zhou M, Dhanasekaran SM, Varambally S, Barrette TR, Sanda MG, et al. alpha-Methylacyl coenzyme A racemase as a tissue biomarker for prostate cancer. JAMA. 2002 3;287(13):1662-70. [ Links ]
*13. True L, Coleman I, Hawley S, Huang CY, Gifford D, Coleman R, et al. A molecular correlate to the Gleason grading system for prostate adenocarcinoma. Proc Natl Acad Sci USA. 2006 18;103(29):10991-6. [ Links ]
14. Petrovics G, Liu A, Shaheduzzaman S, Furusato B, Sun C, Chen Y, et al. Frequent overexpression of ETS-related gene-1 (ERG1) in prostate cancer transcriptome. Oncogene. 2005 26;24(23):3847-52. [ Links ]
15. Hessels D, Klein Gunnewiek JM, van Oort I, Karthaus HF, van Leenders GJ, van Balken B, et al. DD3(PCA3)-based molecular urine analysis for the diagnosis of prostate cancer. Eur Urol. 2003;44(1):8-15; discussion -6. [ Links ]
16. Deras IL, Aubin SM, Blase A, Day JR, Koo S, Partin AW, et al. PCA3: a molecular urine assay for predicting prostate biopsy outcome. J Urol. 2008;179(4):1587-92. [ Links ]
*17. Fradet Y, Saad F, Aprikian A, Dessureault J, Elhilali M, Trudel C, et al. uPM3, a new molecular urine test for the detection of prostate cancer. Urology. 2004;64(2):311-5; discussion 5-6. [ Links ]
18. Marks LS, Fradet Y, Deras IL, Blase A, Mathis J, Aubin SM, et al. PCA3 molecular urine assay for prostate cancer in men undergoing repeat biopsy. Urology. 2007;69(3):532-5. [ Links ]
**19. Tinzl M, Marberger M, Horvath S, Chypre C. DD3PCA3 RNA analysis in urine-- a new perspective for detecting prostate cancer. Eur Urol. 2004;46(2):182-6; discussion 7. [ Links ]
20. Nupponen NN, Visakorpi T. Molecular cytogenetics of prostate cancer. Microsc Res Tech. 2000 1;51(5):456-63. [ Links ]
21. Yano S, Matsuyama H, Matsuda K, Matsumoto H, Yoshihiro S, Naito K. Accuracy of an array comparative genomic hybridization (CGH) technique in detecting DNA copy number aberrations: comparison with conventional CGH and loss of heterozygosity analysis in prostate cancer. Cancer Genet Cytogenet. 2004 15;150(2):122-7. [ Links ]
22. van Duin M, van Marion R, Vissers K, Watson JE, van Weerden WM, Schroder FH, et al. High-resolution array comparative genomic hybridization of chromosome arm 8q: evaluation of genetic progression markers for prostate cancer. Genes Chromosomes Cancer. 2005;44(4):438-49. [ Links ]
*23. Paris PL, Andaya A, Fridlyand J, Jain AN, Weinberg V, Kowbel D, et al. Whole genome scanning identifies genotypes associated with recurrence and metastasis in prostate tumors. Hum Mol Genet. 2004 1;13(13):1303-13. [ Links ]
24. Paris PL, Weinberg V, Simko J, Andaya A, Albo G, Rubin MA, et al. Preliminary evaluation of prostate cancer metastatic risk biomarkers. Int J Biol Markers. 2005;20(3):141-5. [ Links ]
25. Papadopoulos N, Kinzler KW, Vogelstein B. The role of companion diagnostics in the development and use of mutation-targeted cancer therapies. Nat Biotechnol. 2006;24(8):985-95. [ Links ]
26. Narla G, Difeo A, Reeves HL, Schaid DJ, Hirshfeld J, Hod E, et al. A germline DNA polymorphism enhances alternative splicing of the KLF6 tumor suppressor gene and is associated with increased prostate cancer risk. Cancer Res. 2005 15;65(4):1213-22. [ Links ]
27. Huang X, Li X, Guo B. KLF6 induces apoptosis in prostate cancer cells through up-regulation of ATF3. J Biol Chem. 2008 31;283(44):29795-801. [ Links ]
28. Narla G, DiFeo A, Fernandez Y, Dhanasekaran S, Huang F, Sangodkar J, et al. KLF6-SV1 overexpression accelerates human and mouse prostate cancer progression and metastasis. J Clin Invest. 2008;118(8):2711-21. [ Links ]
**29. Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005 28;310(5748):644-8. [ Links ]
*30. Kumar-Sinha C, Tomlins SA, Chinnaiyan AM. Recurrent gene fusions in prostate cancer. Nat Rev Cancer. 2008;8(7):497-511. [ Links ]
31. Hermans KG, van Marion R, van Dekken H, Jenster G, van Weerden WM, Trapman J. TMPRSS2: ERG fusion by translocation or interstitial deletion is highly relevant in androgen-dependent prostate cancer, but is bypassed in late-stage androgen receptor-negative prostate cancer. Cancer Res. 2006 15;66(22):10658-63. [ Links ]
32. Iljin K, Wolf M, Edgren H, Gupta S, Kilpinen S, Skotheim RI, et al. TMPRSS2 fusions with oncogenic ETS factors in prostate cancer involve unbalanced genomic rearrangements and are as sociated with HDAC1 and epigenetic reprogramming. Cancer Res. 2006 1;66(21):10242-6. [ Links ]
33. Soller MJ, Isaksson M, Elfving P, Soller W, Lundgren R, Panagopoulos I. Confirmation of the high frequency of the TMPRSS2/ERG fusion gene in prostate cancer. Genes Chromosomes Cancer. 2006;45(7):717-9. [ Links ]
34. Yoshimoto M, Joshua AM, Chilton-Macneill S, Bayani J, Selvarajah S, Evans AJ, et al. Three-color FISH analysis of TMPRSS2/ERG fusions in prostate cancer indicates that genomic microdeletion of chromosome 21 is associated with rearrangement. Neoplasia. 2006;8(6):465-9. [ Links ]
**35. Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003 20;349(21):2042-54. [ Links ]
36. Kang GH, Lee S, Lee HJ, Hwang KS. Aberrant CpG island hypermethylation of multiple genes in prostate cancer and prostatic intraepithelial neoplasia. J Pathol. 2004;202(2):233-40. [ Links ]
37. Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF, et al. E-cadherin expression is silenced by DNA hypermethylation in human breast and prostate carcinomas. Cancer Res. 1995 15;55(22):5195-9. [ Links ]
38. Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res. 2004 15;64(6):1975-86. [ Links ]
39. Liu L, Yoon JH, Dammann R, Pfeifer GP. Frequent hypermethylation of the RASSF1A gene in prostate cancer. Oncogene. 2002 3;21(44):6835-40. [ Links ]
*40. Chung W, Kwabi-Addo B, Ittmann M, Jelinek J, Shen L, Yu Y, et al. Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling. PLoS ONE. 2008;3(4):e2079. [ Links ]
*41. Ellinger J, Bastian PJ, Jurgan T, Biermann K, Kahl P, Heukamp LC, et al. CpG island hypermethylation at multiple gene sites in diagnosis and prognosis of prostate cancer. Urology. 2008;71(1):161-7. [ Links ]
42. Carpten J, Nupponen N, Isaacs S, Sood R, Robbins C, Xu J, et al. Germline mutations in the ribonuclease L gene in families showing linkage with HPC1. Nat Genet. 2002;30(2):181-4. [ Links ]
43. Tavtigian SV, Simard J, Teng DH, Abtin V, Baumgard M, Beck A, et al. A candidate prostate cancer susceptibility gene at chromosome 17p. Nat Genet. 2001;27(2):172-80. [ Links ]
44. Berthon P, Valeri A, Cohen-Akenine A, Drelon E, Paiss T, Wohr G, et al. Predisposing gene for early-onset prostate cancer, localized on chromosome 1q42.2-43. Am J Hum Genet. 1998;62(6):1416-24. [ Links ]
45. Magee JA, Araki T, Patil S, Ehrig T, True L, Humphrey PA, et al. Expression profiling reveals hep sin overexpression in prostate cancer. Cancer Res. 2001 1;61(15):5692-6. [ Links ]
46. Lin X, Tascilar M, Lee WH, Vles WJ, Lee BH, Veeraswamy R, et al. GSTP1 CpG island hypermethylation is responsible for the absence of GSTP1 expression in human prostate cancer cells. Am J Pathol. 2001;159(5):1815-26. [ Links ]
47. Cordon-Cardo C, Koff A, Drobnjak M, Capodieci P, Osman I, Millard SS, et al. Distinct altered patterns of p27KIP1 gene expression in benign prostatic hyperplasia and prostatic carcinoma. J Natl Cancer Inst. 1998 2;90(17):1284-91. [ Links ]
*48. Yang RM, Naitoh J, Murphy M, Wang HJ, Phillipson J, deKernion JB, et al. Low p27 expression predicts poor disease-free survival in patients with prostate cancer. J Urol. 1998;159(3):941-5. [ Links ]
49. Otto T, Rembrink K, Goepel M, Meyer-Schwickerath M, Rubben H. E-cadherin: a marker for differentiation and invasiveness in prostatic carcinoma. Urol Res. 1993;21(5):359-62. [ Links ]
50. Umbas R, Isaacs WB, Bringuier PP, Schaafsma HE, Karthaus HF, Oosterhof GO, et al. Decreased E-cadherin expression is associated with poor prognosis in patients with prostate cancer. Cancer Res. 1994 15;54(14):3929-33. [ Links ]
51. Umbas R, Schalken JA, Aalders TW, Carter BS, Karthaus HF, Schaafsma HE, et al. Expression of the cellular adhesion molecule E-cadherin is reduced or absent in high-grade prostate cancer. Cancer Res. 1992 15;52(18):5104-9. [ Links ]
52. Bowen C, Bubendorf L, Voeller HJ, Slack R, Willi N, Sauter G, et al. Loss of NKX3.1 expression in human prostate cancers correlates with tumor progression. Cancer Res. 2000 1;60(21):6111-5. [ Links ]
53. Webster R. Filamentous phage biology. In: Barbas CF, 3rd; Burton, D. R.; Scott, J. K.; Silverman, editor. Phage Display: A Laboratory Manual: Cold Spring Harbor Laboratory Press; 2001. p. 1.-.37. [ Links ]
54. Scott JK, Smith GP. Searching for peptide ligands with an epitope library. Science. 1990 27;249(4967):386-90. [ Links ]
55. Smith GP, Scott JK. Libraries of peptides and proteins displayed on filamentous phage. Methods Enzymol. 1993;217:228-57. [ Links ]
56. Arap MA. Phage display technology - Applications and innovations. Genetics and Molecular Biology. 2005;28(1):1-9. [ Links ]
57. Koivunen E, Arap W, Rajotte D, Lahdenranta J, Pasqualini R. Identification of receptor ligands with phage display peptide libraries. J Nucl Med. 1999;40(5):883-8. [ Links ]
*58. Mintz PJ, Kim J, Do KA, Wang X, Zinner RG, Cristofanilli M, et al. Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat Biotechnol. 2003;21(1):57-63. [ Links ]
59. Lee AS. Mammalian stress response: induction of the glucose-regulated protein family. Curr Opin Cell Biol. 1992;4(2):267-73. [ Links ]
60. Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. 1993 5;259(5100):1409-10. [ Links ]
61. Munro S, Pelham HR. An Hsp70-like protein in the ER: identity with the 78 kd glucose-regulated protein and immunoglobulin heavy chain binding protein. Cell. 1986 18;46(2):291-300. [ Links ]
62. Melnick J, Argon Y. Molecular chaperones and the biosynthesis of antigen receptors. Immunol Today. 1995;16(5):243-50. [ Links ]
63. Li WW, Alexandre S, Cao X, Lee AS. Transactivation of the grp78 promoter by Ca2+ depletion. A comparative analysis with A23187 and the endoplasmic reticulum Ca(2+)-ATPase inhibitor thapsigargin. J Biol Chem. 1993 5;268(16):12003-9. [ Links ]
64. Jamora C, Dennert G, Lee AS. Inhibition of tumor progression by suppression of stress protein GRP78/BiP induction in fibrosarcoma B/C10ME. Proc Natl Acad Sci U S A. 1996 23;93(15):7690-4. [ Links ]
65. Sugawara S, Takeda K, Lee A, Dennert G. Suppression of stress protein GRP78 induction in tumor B/C10ME eliminates resistance to cell mediated cytotoxicity. Cancer Res. 1993 15;53(24):6001-5. [ Links ]
66. Miyake H, Hara I, Arakawa S, Kamidono S. Stress protein GRP78 prevents apoptosis induced by calcium ionophore, ionomycin, but not by glycosylation inhibitor, tunicamycin, in human prostate cancer cells. J Cell Biochem. 2000;77(3):396-408. [ Links ]
*67. Arap MA, Lahdenranta J, Mintz PJ, Hajitou A, Sarkis AS, Arap W, et al. Cell surface expression of the stress response chaperone GRP78 enables tumor targeting by circulating ligands. Cancer Cell. 2004;6(3):275-84. [ Links ]
68 Arap W, Haedicke W, Bernasconi M, Kain R, Rajotte D, Krajewski S, et al. Targeting the prostate for destruction through a vascular address. Proc Natl Acad Sci U S A. 2002 5;99(3):1527-31. [ Links ]
69. Ellerby HM, Arap W, Ellerby LM, Kain R, Andrusiak R, Rio GD, et al. Anti-cancer activity of targeted pro-apoptotic peptides. Nat Med. 1999;5(9):1032-8. [ Links ]
70. Hajitou A, Sounni NE, Devy L, Grignet-Debrus C, Lewalle JM, Li H, et al. Down-regulation of vascular endothelial growth factor by tissue inhibitor of metalloproteinase-2: effect on in vivo mammary tumor growth and angiogenesis. Cancer Res. 2001 15;61(8):3450-7. [ Links ]
**71. Lebret T, Watson RW, Fitzpatrick JM. Heat shock proteins: their role in urological tumors. J Urol. 2003;169(1):338-46. [ Links ]
72. Liu C, Bhattacharjee G, Boisvert W, Dilley R, Edgington T. In vivo interrogation of the molecular display of atherosclerotic lesion surfaces. Am J Pathol. 2003;163(5):1859-71. [ Links ]
73. Petricoin EF, 3rd, Ornstein DK, Paweletz CP, Ardekani A, Hackett PS, Hitt BA, et al. Serum proteomic patterns for detection of prostate cancer. J Natl Cancer Inst. 2002 16;94(20):1576-8. [ Links ]
 Dirección para correspondencia:
Dirección para correspondencia:
Marco Antonio Arap, MD. PhD.
Rua Guarara 329 apto 101
01425-001 São Paulo. SP. (Brazil)
marcoarap@hotmail.com
Aceptado para publicar: 1 de julio 2009.

