










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO  Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista de Diagnóstico Biológico
versión impresa ISSN 0034-7973
Rev Diagn Biol vol.52 no.1 ene./mar. 2003
GRUPOS DE TRABAJO AEBM
Metodología para la estimación del error preanalítico y su significación, en determinaciones realizadas a partir de especímenes obtenidos en puntos periféricos de obtención y recogida de especímenes (PPORE)
Dr. Santiago Prieto1. Dra. Carmen Sempere2. Dra. M. Luisa Salve3. Dr. J Manuel Moreno4.
Servicio de Análisis Clínicos: 1 Hospital Virgen de la Luz. Cuenca 2 Hospital de Sagunto. Valencia 3 Hospital de La Plana. Villareal. Castellón 4 Fundación Hospital Alcorcón.
Palabras clave: Garantía de calidad, preanalítica.
Key Words: Quality Assurance, preanalytical error.
RECOMENDACION elaborada a partir de la presentación realizada en el X CONGRESO DEL LABORATORIO CLINICO (MURCIA MAYO 2001) con el titulo PROTOCOLOS DE VALlDACIÓN DE PRUEBAS EN PUNTOS PERIFERICOS DE OBTENCIÓN Y RECOGIDA DE ESPECIMENES
Recibido: 4-IX-02
Aceptado: 27-XII-02
Correspondencia: Dr. Santiago Prieto.
Servicio de Análisis Clínicos. Hospital Virgen de la Luz.
c/Donantes de sangre s/n Cuenca 16002.
|
"Prefiero debatirme en la incertidumbre que acomodarme en el error"
1.- Introducción
Este documento se enmarca dentro de los generados por la AEBM (Asociación Española de Biopatología Médica) relativos a los puntos periféricos de obtención y recogida de especímenes (PPORE).
En el documento Problemática de los Puntos de Extracción Periféricos de los Laboratorio de Análisis Clínicos1 se aborda una visión general y una revisión del tema a nivel europeo y de las CCAA.
Queda claro que nuestra postura genérica no es favorable de entrada a los mismos. Son una opción aceptable cuando no existe otra alternativa mejor y siempre que se tomen las medidas necesarias para garantizar la calidad preanalítica en estos centros.
En el documento Garantía de calidad de la fase preanalítica extralaboratorio en puntos periféricos de obtención y recogida de especímenes 2 se plantean aspectos de definición y de organización de dichos puntos para que desde el laboratorio pueda realizarse un adecuado control que garantice que las actividades adicionales que genera la extracción periférica no incrementan el error preanalítico más allá de los límites establecidos por el laboratorio
En este documento se plantea proponer al especialista de laboratorio una metodología estandarizada que le permita decidir, en caso de existencia de dichos puntos, si la realización de una prueba concreta puede asignarse a la cartera de servicios de un punto concreto. Cómo medir el error y cuales son los criterios de tolerancia que podemos definir.
Esta metodología debe posibilitar la definición de las carteras de servicios de puntos concretos para los que no exista evidencia suficiente que permita asignar o rechazar una prueba específica a dicha cartera.
Debemos insistir en la diferencia entre espécimen y muestra.
El espécimen esta tomado directamente del paciente, la muestra es una parte o todo el espécimen manipulado para aumentar la estabilidad o facilitar su manejo en el análisis.
El espécimen no lleva preparación o aquella es mínima; por lo tanto no requiere recursos adicionales a los de la propia extracción, pero su estabilidad es menor que la de la muestra, y en el caso de analitos distintos que requieran una conservación distinta (por ej. distintas temperaturas: en el caso del hemograma es distinto si queremos realizar contaje o formula leucocitaria) hay que elegir una de ellas ( o sacar uno o más especímenes adicionales).
La muestra requiere un procesamiento y unas condiciones de almacenaje específicas para cada test o grupo de test solicitados; requiere por lo tanto recursos adicionales a los de la propia extracción, incluyendo personal formado específicamente y más tiempo de procesamiento. Por contra la estabilidad es mayor que en el caso del espécimen.
No es lo mismo transportar especímenes que muestras.
Si bien las reflexiones y recomendaciones metodológicas aquí citadas se refieren en primer lugar a los PPORE por su mayor riesgo teórico, no debe olvidarse que las extracciones en plantas o consultas del centro hospitalario, si no están igualmente controladas puede originar también el mismo tipo de errores.
Desde este punto de vista, la metodología citada puede ser utilizada también en dichos ámbitos cuando se sospeche que existen algunos de los elementos de distorsión citados (temperatura, tiempo, efectos mecánicos, etc.).
2.- La extracción periférica tutelada por el laboratorio.
La calidad clínica de los puntos de extracción pasa porque estén tutelados por el laboratorio del que dependen. El facultativo que firma el informe, asume los errores cometidos en su elaboración. Por eso es la persona capacitada para diseñar, hacer cumplir y controlar el sistema de aseguramiento de la calidad (esquemas de garantia y control de calidad). Esa responsabilidad no es delegable y no asumirla es una negligencia inexcusable.
En la figura 1 se exponen en forma de gráfico de espina de pescado los principales elementos que pueden actuar como fuente potencial de error en la fase preanalítica extralaboratorio.
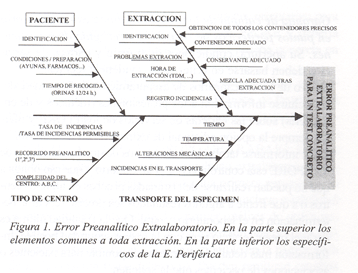
Dentro de ese esquema global se incluyen los PPORE, que además añaden características propias (ver parte inferior del esquema de la fig. 1) .
Cada laboratorio debe definir:
* Las condiciones de obtención de los especímenes y su procesamiento extralaboratorio.
* Las condiciones para recepcionar los especímenes y muestras remitidos desde los PPORE.
* Los mecanismos de control y seguimiento de errores.
Las NORMAS AEBM 98 3 en su punto 6.1 (EXTRACCION PERIFERICA DE ESPECIMENES / MUESTRAS) indican que:
" El laboratorio es responsable de la calidad de los informes que emite. Dicha calidad es un proceso global, del que un aspecto significativo es la obtención y procesamiento preanalítico.
El laboratorio debe controlar ese proceso. Si existen dudas sobre la calidad de la obtención, el transporte, procesamiento o identificación de la muestra, ésta debe ser rechazada.
Si un punto concreto de extracción se identifica en auditorias como una potencial fuente de no conformidades, deberían realizarse acciones correctoras que van desde la formación del personal y revisión del sistema de aseguramiento de la calidad de dicho punto hasta el cierre temporal o definitivo del mismo."
E1 70-80 % de los errores del laboratorio en el momento actual se encuentran situados en la fase preanalítica4. ¿Cual puede ser la aportación de una fase preanalítica sin control al aseguramiento de la calidad en el laboratorio clínico? Indudablemente un desastre desde todos los puntos de vista.
La Extracción Periférica puede incrementar el error preanalítico (disminuir la calidad). Por eso es una alternativa cuando no hay otra opción posible. La pregunta en estos casos es doble:
1) ¿Existe una alternativa que suministre la misma o semejante información clínica obviando los riesgos de error para una prueba concreta en un PPORE?
2) ¿Cual es el límite de error que podemos permitirnos dentro de los PPORE, de manera que dicho error no exista o al menos no sea significativo al interpretar el dato?
Las respuestas vendrán mediadas en función de ...
* La accesibilidad alternativa: Debemos entender y defender que el paciente no tiene obligatoriamente que acudir a su PPORE. Si existe es una opción, pero no una obligación. ¿Puede el paciente acudir al laboratorio en otro momento? El paciente puede estar citado en el centro de especialidades u hospital para otra prueba o interconsulta.
* ¿existen pruebas alternativas a la solicitada que puedan dar semejante información clínica? En recorridos preanalíticos mayores de 3 horas, la utilidad del sedimento urinario es cuestionable5 en el estudio de las células, pero la tira reactiva puede dar información útil y a menudo suficiente.
* en función del tiempo de respuesta que es necesario para el clínico.
- a menudo se solicitan análisis que van a ser valorados semanas después...probablemente el paciente podrá acudir al laboratorio durante ese periodo.
- o bien el clínico precisa los resultados con celeridad y el punto de extracción tiene una frecuencia de una vez por semana. Debe existir la opción de que acuda al laboratorio y disponga de los resultados de manera rápida.
* en función de si se trata de resultados (valores) de orden clínico, diagnóstico, epidemiológico o de investigación.
- Ej 1: un determinado PPORE puede tener un bias negativo para la glucosa de unos 7-10 mg/dl. Podemos darlo por bueno para seguimientos de diabéticos, pero no para establecer diagnósticos según los nuevos criterios del ADA. Si lo que se está valorando es un proyecto de investigación, el criterio debe ser mucho mas estricto aún.
- Ej 2: un determinado PPORE presenta una cierta frecuencia de muestras hemolizadas y valores de potasio con un bias positivo de 0.2-0.3 mEq/l. Podemos aceptarlo con reservas para monitorización de tratamiento con diuréticos ahorradores de potasio, pero no para diuréticos tiazídicos o dentro del protocolo de hipertensión donde usamos como screening de hiperaldosteronismo la hipopotasemia.
El objetivo no es que un porcentaje de resultados en sujetos normales puedan ser válidos, sino que la posibilidad de error en cualquier tipo de paciente para cualquiera de las concentraciones que pueda presentar en esa prueba sea inferior al limite de tolerancia establecido por el laboratorio.
En resumen, los puntos de extracción periférica, si existen, deben estar tutelados, organizados y controlados por el laboratorio del que dependen. Deberían clasificarse y organizarse según los criterios descritos en el documento de AEBM
Garantía de calidad de la fase preanalítica extralaboratorio en puntos periféricos de obtención y recogida de especímenes. Su apertura, condiciones de trabajo y cartera de servicios deben basarse en criterios de evidencia técnica.
Dentro de los mecanismos de aseguramiento de la calidad, debe incluirse información a los profesionales (médicos y de enfermería) sobre las carteras de servicios de los PPORE y ofertar siempre la opción adicional de extracción en laboratorio.
Debe informarse también a los usuarios de que la extracción de los PPORE esta controlada por el laboratorio y que eso obliga a que no puedan realizarse determinadas pruebas en algunos centros o a que frente a algunos resultados sea preciso repetir la determinación en el laboratorio central. Que la finalidad última es darle el mejor servicio pero con la mayor seguridad posible. Información más detallada debe estar disponible para pacientes o asociaciones de pacientes que la soliciten.
La extracción periférica no debe ser la base sino un complemento entendido como servicio al paciente si no existe otra alternativa y para unas pruebas concretas.
Desde la perspectiva de la calidad un error detectado es una oportunidad de mejora, pero un error ignorado es un acto negligente y contrario a dicha practica de la calidad. Un acto contrario a la ética que debe ser inherente a la práctica de nuestra especialidad.
¿Cómo definir la cartera de servicios de cada punto?
La cartera de servicios de un PPORE estaría compuesta por aquellas pruebas cuya realización al añadir el incremento de error de la fase preanalítica extralaboratorio (Ver figura 1 parte inferior) no se vieran afectadas desde el punto de vista del uso que fuera a hacerse de su interpretación.
¿Cuáles son los mecanismos de error que aporta la extracción periférica al laboratorio? Básicamente cuatro:
* Tres de ellos son variables continuas (el error se incrementa al incrementarse la magnitud de la variable pero no existe un punto de corte critico o sólo se da en casos muy extremos). Las variables son:
Temperatura (definirla y monitorizarla)
Tiempo (definido como recorrido preanalítico)
Efectos mecánicos (el tipo de vehiculo y la ruta deben ser siempre los mismos)
El peso de cada variable a su vez dependerá de la prueba de que se trate, del spectrum bias o población de estudio (a) y de si la solicitud es para diagnóstico o seguimiento.
* Una cuarta variable, a menudo infravalorada es el conocimiento/desconocimiento know how). La gestión de esa variable por los profesionales que trabajan en el PPORE es critica para obtener unos resultados fiables. De nuevo la formación, control y evaluación del desempeño, deben estar tutelados por el laboratorio.
Debería existir una base de datos de carácter orientativo para definir las pruebas del catalogo de cada PPORE y debería existir un protocolo de validación de una prueba concreta en un PPORE concreto.
En el primer caso podemos partir de la bibliografía y más específicamente en nuestro medio del Catálogo de Pruebas del INSALUD 5, consensuado entre todas las asociaciones científicas del área de laboratorio.
- OBJETIVO 1 DE ESTE GRUPO DE TRABAJO: Diseñar una base de datos sobre error preanalítico extralaboratorio relacionado con E. Periférica. Como una primera aproximación se cita el catálogo del INSALUD 1999 que incluye estabilidad de especímenes y muestras. Un primer análisis de orden general se incluye en la tabla I . Se ha realizado una primera versión de la base de datos que esta disponible en la página Web de la Asociación (vww.aebm.org). Pueden remitirse estudios de estabilidad de analitos (ver anexo 2) que serán actualizados en dicha base de datos.

- OBJETIVO 2 DE ESTE GRUPO DE TRABAJO: Definir una metodología/s para validación de test concretos.
Ante la definición de la cartera de servicios de un centro, el analista debería
(1) consultar la estabilidad para el recorrido preanalítico que define a dicho centro
(2) si no encuentra dicha información o sus condiciones no están descritas, validar la técnica en su caso y
(3) al ser normalizada la metodología, poder remitir sus datos para incrementar la información y la potencia estadística de la citada base de datos.
3.- Metodología
En primer lograr debemos hacer constar que en caso del ensayo "pre" lo adecuado es obtener de los pacientes a los que se les va a extraer un volumen mayor de sangre del necesario. Se hace precisa su autorización (consentimiento) que puede ser verbal o escrito.
Esto no es necesario en el ensayo "post", ya que se utilizan los datos que han sido utilizados en el proceso clínico y en el tratamiento estadístico se eliminaran las referencias de identificación del paciente.
3.1 Modelos de protocolo
Se definen dos:
* Ensayo "pre": para aplicar antes de dar de alta una prueba en un PPORE.
* Ensayo "post": para validar pruebas que ya estén en marcha y no se tenga garantía bibliográfica de ausencia de error o como prueba confirmatoria ante un ensayo pre en el limite de tolerancia que se haya decidido utilizar en periodo de prueba durante un tiempo.
3.1.1 Ensayo de simulación con datos pareados (ensayo pre)
Se trata de un ensayo de tipo prospectivo: especímenes extraídos simultáneamente y procesados uno en condiciones normales y el otro/s transportado en un vehículo como los utilizados en la extracción periférica durante el tiempo máximo que se va a conceder a dicho PPORE incrementado en 15 minutos (por ejemplo si su recorrido preanalítico está en dos horas, debe circular durante 2 horas 15 min antes de acudir para su procesamiento) b
I).- Recogida de especímenes
Recoger especímenes de pacientes que acudan a la toma de muestras del laboratorio central, sin tener en cuenta su estado clínico para así obtener intervalos de concentraciones lo más amplio posible. Obtener los especímenes adecuados en cada caso:
* 3 tubos para la obtención de suero (estudio de la estabilidad de metabolitos en suero). A, B y C.
* 3 tubos para la obtención de plasma (estudio de la estabilidad de metabolitos en plasma). A, B y C.
* 2 tubos de sangre total (estudio de la estabilidad de los elementos formes de la sangre). A y B.
* 2 tubos de sangre total (estudio de la estabilidad de la medición de la eritrosedimentación). A y B.
* 2 alícuotas de orina de la primera de la mañana para el estudio de la estabilidad de los resultados de su análisis (tira reactiva + sedimento urinario + otros constituyentes). A y B.
II).- Manejo de especímenes
Los especímenes obtenidos de los pacientes son sometidos a distintos tratamientos con el fin de estudiar si existen diferencias significativas entre los especímenes obtenidos en el laboratorio (tubo A) y los obtenidos en los puntos periféricos, y enviados al laboratorio sin centrifugar (tubo B) o centrifugados (tubo C).
TUBO A.- Los especímenes y alícuotas de este grupo se procesan según el protocolo habitual de nuestro laboratorio, teniendo en cuenta que esta circunstancia se empleará como estandar (valor verdadero) para los estudios de estabilidad. El laboratorio debe tener protocolizados todos los procesos preanalíticos intralaboratorio según las indicaciones propuestas en bibliografías autorizadas.
TUBO B.- Los especímenes de este grupo se conservan en la nevera portátil refrigerada de la misma forma en que vamos a llevar a cabo el transporte (seria recomendable tener una nevera en la que podamos registrar la temperatura en su interior). Se transportan por un circuito que simule el trayecto original y durante el tiempo fijado (recorrido preanalítico). A su vuelta al laboratorio se tratan según los protocolos del laboratorio, dejando registradas la hora de llegada y la hora de centrifugación en aquellos especímenes en los que sea necesaria esta actuación .
TUBO C.- Los especímenes de este grupo son centrifugados tras la extracción, al igual que los del grupo A, y luego sometidos al mismo tratamiento que los del grupo B.
III)- Análisis de los especímenes
Los metabolitos o elementos formes que queremos validar de los especímenes sometidos a los procedimientos anteriormente descritos deben ser analizados de forma duplicada para evitar los errores aleatorios que puedan ocurrir durante el análisis y a ser posible los del grupo B y C deberían realizarse en la misma serie analítica para evitar en lo posible las variaciones interserie.
3.1.2 Ensayo retrospectivo con datos históricos (ensayo post)
El modelo de ensayo es procesar un número elevado de pacientes "homogeneos" cuyos especímenes hayan sido recibidos por extracción normal o periférica. Se adjunta como anexo 1 un ejemplo 6.

Se obtendrá por extracción de la base de datos del SIL.
El número de pacientes será lo más elevado posible.
La homogeneidad se consigue a igualdad de diagnósticos o programas de Atención Primaria, o si no se dispone de esa información utilizando orígenes semejantes: por ejemplo sólo pacientes de Atención Primaria, o pacientes de la misma consulta de especialidad (debe ponerse atención en no mezclar consultas monográficas con generales).
El periodo de tiempo de recogida debe ser el mismo o semejante (no comparar datos de meses distintos) para evitar sesgos de cambios estacionales en el parámetro o tipo de paciente (algunos programas de Atención Primaria captan más pacientes en una época del año, etc.) o cambios en la organización o personal que realiza las distintas tareas (el personal en verano será más eventual que durante el resto del año, etc.).
3.2 Muestra
¿Cuántas muestras o pacientes deben procesarse en cada caso?
El tamaño muestral podemos deducirlo a partir del intervalo de confianza de la media de las diferencias: debe ser tal que aunque tomemos los extremos de dicho intervalo, la decisión sea la misma. O dicho de otra manera, si uno de 1os extremos del intervalo es válido y el otro no, debemos aumentar el tamaño muestral para reducir dicho intervalo.
Esto es critico en el ensayo pre donde vamos a realizar determinaciones adicionales. En el ensayo post, como procesaremos nuestra base de datos se propone partir de un número lo más elevado posible para aumentar la potencia estadística.
En el ensayo pre se recomienda, sobre todo si es una prueba de alto coste, hacer un ensayo piloto con 10-30 datos para definir a partir de él el tamaño muestral. Si es una prueba de bajo coste, puede realizarse con 100-200 datos de entrada que en general serán suficientes.
¿Debe hacerse para distintos niveles o tipos de pacientes?
En base a las características de la prueba [el ejemplo del tiempo de protrombina en pacientes teóricamente sanos o en tratamiento con anticoagulantes orales, o el análisis de PTH en pacientes teóricamente sanos o en pacientes en IRC (Insuficiencia Renal Crónica), etc.] o bien a partir de una primera inspección de los resultados puede esperarse o sospecharse que intervalos distintos o grupos de pacientes distintos se comporten de manera diferente. En ese caso, además del ensayo global, debe diseñarse uno especifico.
3.3. Limite de tolerancia
Una vez definido el tipo de ensayo y la muestra debemos, antes de proceder al estudio, definir el limite de tolerancia que vamos a considerar.
Se define como limite de tolerancia el valor máximo de error (en control de calidad industrial de error de dispersión) por debajo del cual, no es necesario tomar medidas sobre el proceso 7.
¿Qué elementos podemos utilizar para definir el limite de tolerancia?
En nuestro control de calidad identificamos un error de inexactitud (diferencia entre nuestra media y la media diana) y error de dispersión (desviación estándar). Estas dos variables a su vez pueden definirse intraensayo, interensayo a corto plazo (por ejemplo para los valores acumulados de un mes) e interensayo a largo plazo (por ejemplo para los valores acumulados de un semestre o de un año).
Podemos incluir en un único dato los valores de inexactitud y dispersión. Seria el error máximo tolerable (TEa) (8):
TEa = (inexactitud) + Z (imprecisión)
La inexactitud se define a partir del bias y el SE (SE= error sistemático a largo plazo) La imprecisión a partir del error aleatorio (RE= error aleatorio a largo plazo) Z se define en función del número de desviaciones estándar (1.65, 1.96, 2.33)
En el caso que nos ocupa, debemos definir para cada analito unos limites de tolerancia en el bias y en la SD que nos permitan expresar que el incremento de error de la fase preanalítica extralaboratorio no llega a un nivel de error clínico.
Esos limites pueden ser únicos para el analito o distintos en función del rango de medida(concentración).
Podemos aceptar los limites de la bibliografia 9, 10 citando las referencias de donde las tomamos o calcular con la fórmula indicada nuestro error máximo a partir de los valores de imprecisión e inexactitud de nuestros propios datos.
Aunque en una primera aproximación parece razonable utilizar el CV biológico (de hecho proponemos usar SD y en los objetivos de control de calidad una aproximación recomendada en la bibliografía al uso es usar 1/4 o 1/2 del CV biológico como objetivo de calidad), no parece adecuado utilizarlo en este caso por:
* Si bien es una recomendación de distintos grupos, no es uniforme, ni los datos del CV Biológico lo son en todos los estudios, ni existen datos de todos los parámetros y además el CV Biológico se define en valores "de referencia", pero no en niveles patológicos. Por otro lado existen parámetros donde el CV Biológico se considera muy estricto y se acepta el estado del arte (iones) y otros en que es muy amplio y se prefiere ser aún mas restrictivo (algunos enzimas).Es decir el limite de tolerancia no seria universalmente aceptado sino plantearía una variabilidad
* La hipótesis nula como hipótesis de trabajo: la base de nuestro razonamiento es que (en el caso del ensayo pre) una muestra del mismo paciente, obtenida en el mismo instante, debe dar idéntico resultado (salvo que el procesamiento preanalítico genere alguna variación). Por lo tanto la variación admitida debe ser la de duplicados intraensayo/interensayo medidos en el laboratorio.
* Queremos comprobar si dicha hipótesis se cumple EN LAS CONDICIONES DE TRABAJO DEL LABORATORIO, por lo que no podemos tomar una desviación estandar de la literatura, sino la nuestra (otra cosa es que nuestros objetivos de calidad sean los del CV biológico en una fracción concreta y además los cumplamos con exactitud (ni en más ni en menos).
* Deberíamos pues cumplir los criterios de interensayo intra dia. Como eso debería calcularse, de ahí la propuesta de interensayo en tiempo corto (1 mes) que es una opción mas que razonable para evidenciar diferencias si existen.
Propuesta concreta (aunque no excluyente) para el documento actual
Los valores base de inexactiud e imprecisión que se proponen son los del QC acumulado del último mes. Como control adicional podemos comprobar dicho valor mes a mes los tres últimos meses para comprobar que no se ha producido ninguna modificación sustantiva en la metrología del analito c.
Dar como tolerancia el error que aceptamos acumulado en un mes cuando la comparación la vamos a hacer intra día, parece suficiente margen . Puede completarse con criterios clínicos para algunos analitos. El valor critico se situaría en principio entre 2 y 3 veces el valor de la SD.
3.4 Control interno
Siempre que sea posible, es bueno utilizar algún tipo de control interno que nos asegure homogeneidad en la muestras procesadas por extracción periférica y por vía normal.
En el área de la bioquímica, el colesterol total es estable en situaciones de temperatura, tiempo y efectos mecánicos. La demostración de no diferencias en el grupo estudiado para colesterol total unido a diferencias en el analito a valorar, refuerza los resultados del ensayo de comparación.
Seria deseable la búsqueda de otros analitos de comportamiento semejante para muestras de sangre total y orina....
3.5 Análisis estadístico e interpretación de resultados
Las preguntas a responder son:
(1) ¿Existe diferencia entre los valores a nivel de exactitud y/o precisión?
(2) Esa diferencia si existe ¿,invalida el uso de la prueba a todos los niveles o sólo en algunos escenarios o características?
3.5.1 Inspección visual de los resultados 11
Debe realizarse en primer lugar una representación gráfica
* Gráfico X/Y
* Gráfico de Altman Bland 12
Por la inspección visual ya podemos hacernos una idea de si son distribuciones semejantes, de si existe algún nivel que se comporta de manera distinta, etc
3.5.2 Estadística básica
Las distribuciones deberían estudiarse como media (media e intervalo de confianza de la media), SD, CV y distribución en cuartiles (P25, P50, P75, valores mínimo y máximo).
3.5.3 Tests estadísticos a utilizar en el caso del ensayo pre
* para variables continuas se usa la t -Student para datos apareados. Se trata en teoria (hipótesis nula) de datos apareados y que deben tratarse como tales, por lo que si no existen diferencias, la distribución tiene que ser normal y el grado de diferencia estadística según la t nos indica si las diferencias son estadísticamente significativas. Con el criterio del limite de tolerancia ajustamos además dicha diferencia a nuestra escenario de trabajo real.
* en el caso de variables semicuantitativas (variables discretas) T de Wilcoxon para datos apareados: Prueba no paramétrica para variables que no cumplen los supuestos de normalidad, o bien es difícil comprobarlos porque son muestras pequeñas, o para variables semicuantitativas.
En el caso de ensayo post utilizaríamos una t-Student (analizando previamente si existe homogeneidad de varianzas o no con el test F) o una T Wilcoxon para datos apareados en función del tipo de variable.
En la interpretación de los resultados, además del valor del estadístico (expresado como p e intervalo de confianza) deberíamos valorar comparación intercuartiles (valor de las diferencias).
Las diferencias se interpretarían en función de los valores de Z ( Z= valor de la diferencia entre medias/ /SD) es decir, como el número de veces que la diferencia representa el valor de la SD.
* Valores < 1 Z serian considerados idénticos
* Valores 1-2 Z serian considerados como aceptables
* Valores entre 2-3 Z deberían ser valorados a la luz de criterios clínicos o en función de la información aportada por la variabilidad biológica
* Valores > 3Z serian considerados inaceptables
En caso del intervalo de confianza de la media y de la media de las diferencias (en la t-Student), si el rango mínimo es menor de 2-3 Z, pero el rango máximo es mayor, es necesario aumentar el tamaño de la muestra.
Conviene valorar además en el caso de distribuciones normales si existe diferencia entre sus varianzas mediante el test F de Snedecor. ¿Porqué? Porque en el caso de que no haya diferencia de medias pero si de varianzas, habría que tener el dato en consideración y buscar una explicación (¿,otros elementos de dispersión como hemólisis o pretratamiento que afecta a unas muestras si y otras no, etc.?).
3.5.4 Notas al tratamiento estadístico (ensayo pre) 13-16
Primero se observa que no existan diferencias entre los resultados duplicados obtenidos, rechazando del estudio los resultados diferentes y cogiendo la media de las dos determinaciones como resultado
Para estudiar las diferencias existentes entre los distintos especímenes sometidos a diferentes tratamientos se comparan entre si las medias de los resultados obtenidos en cada uno de los grupos mediante los siguientes test estadísticos:
a) t de Student para datos apareados y muestras grandes
(n>30): Cuando estamos comparando variables cuantitativas continuas (por ejemplo concentración de glucosa en sangre). Si la diferencia es estadísticamente significativa debemos establecer el intervalo de confianza de esta diferencia con la formula:

b) T de Wilcoxon para datos apareados: Prueba no paramétrica para variables que no cumplen los supuestos de normalidad, o bien es difícil comprobarlos porque son muestras pequeñas, o para variables semicuantitativas. A cada resultado se le asigna una categoría cuantitativa discreta (por ejemplo, el resultado de la tira reactiva para análisis de orina para el constituyente nítritos cuando es negativo categoría 1 y cuando es positivo categoría 2)
Los pares resultantes del estudio son:
Grupo A - B: Se comparará el par formado por los especímenes procesados según el protocolo habitual del laboratorio, y los especímenes sometidos a transporte sin centrifugar.
Grupo A - C: se comparará el par formado por los especímenes procesados según el protocolo habitual del laboratorio, y los especímenes centrifugados antes de ser sometidos a transporte.
Grupo B - C se comparará el par formado por los especímenes sometidos a transporte, con o sin centrifugación previa.
Para los pares de datos en los que existen diferencias estadísticas significativas debemos proseguir el estudio y averiguar si estas diferencias son igualmente significativas desde el punto analítico y/o clínico.
3.5.5 Hipótesis. y esquema de toma de decisiones
En resumen, debemos establecer que si no existen diferencias de ningún tipo, la prueba puede ser realizada en el PPORE en las condiciones en las que se ha valorado.
Si las diferencias son estadísticas pero no analíticamente significativas, podemos utilizarla aunque indicando que en determinados ámbitos (estudios epidemiológicos o de investigación) los resultados deben ser valorados teniendo en cuenta la incertidumbre adicional. Si se trata de un ensayo pre, seria bueno diseñar un ensayo post para comprobar si la rutina habitual incrementa ese sesgo.
Si las diferencias son analíticamente significativas (superan el limite de tolerancia que hemos definido), no podemos utilizar dicha prueba o bien podemos utilizarla de manera restringida (como screening o prueba orientativa) si su uso puede mejorar la accesibilidad en un número elevado de pacientes (por ejemplo en recorridos preanalíticos mayores de 3 horas no debería realizarse el sedimento, pero si la detección por química seca de nitratos, hemoglobina y esterasa leucocitaria. Un valor negativo excluiría la necesidad de acudir al laboratorio para hacer el sedimento).
Si las diferencias son además clínicamente significativas (superan los criterios clínicos definidos d ), no puede realizarse la determinación en ningún caso, porque inducirá a error en la valoración que del paciente hace el médico.
4.- Generación de información
De todo lo anterior debemos definir un modelo de informe final que sea uniforme para
- que todos podamos entenderlo rápida y fácilmente
- que pueda ser integrado en una base de datos común
- que permita a posteriori estudios de tipo meta-análisis
- Debería recolectarse esa información en una base de datos bibliográfica sobre estabilidad de analitos en determinadas circunstancias (sobre todo de especímenes)
4.1 Modelo de informe
4.2 Información que debe recogerse para la base de datos
A) Identificación del grupo que realiza el estudio. Referencia bibliográfica si ha sido publicado.
B) Identificación del analito, especimen, manipulación.
- B1 Analito
- B2 Unidades. Valores de referencia.
- B3 Método/Analizador
- B4 Código (por ejemplo catálogo INSALUD)
- B5 Espécimen/Muestra, contenedor y manipulación
- B6 Recorrido preanalítico
- B7 Temperatura
- B8 Valoración de efectos mecánicos (en función del tipo de transporte, estado de las carreteras, distancia a recorrer por el transportista a mano con las neveras): Mínimos, Medios, Máximos (tabla 2).

C) Descripción del estudio
- C I Tipo de ensayo. Cómo se ha garantizado la homogeneidad de la población estudiada
- C2 Bias de referencia (el propio en el mes valorado)
- C3 SD ( el propio en el mes valorado)
- C4 Límites de tolerancia fijados
- C5 Existencia de control interno (qué prueba y resultados)
- C6 Resultados:
- Media de los dos conjuntos de datos (normal y PPORE) con su intervalo de confianza
- Valores mínimos de los dos conjuntos de datos (normal y PPORE)
- P25 de los dos conjuntos de datos (normal y PPORE)
- P50 de los dos conjuntos de datos (normal y PPORE)
- P75 de los dos conjuntos de datos (normal y PPORE)
- Valores máximos de los dos conjuntos de datos (normal y PPORE)
- Idem los anteriores pero para las diferencias.
- C7 Técnicas estadísticas utilizadas
- Descripción
- Valor de la p/intervalo de confianza
- Otros resultados

D) Valoración por parte del grupo investigador de los resultados para ese analito en las condiciones expresadas.
Tres opciones:
- (1)Valido
- (2)cuestionable/no concluyente/sólo en algunas circunstancias
- (3)no válido
4.2.2 Actualización
La idea es que los distintos componentes de la comunidad científica del laboratorio puedan remitir sus datos con la estructura anterior. Tras su revisión se integraría en la base de datos para que pudiera ser consultada por otros profesionales y servir de elemento de evidencia para fijar los catálogos de servicios de los PPORE. Los datos remitidos se revisarían e integrarían en la base de datos con indicación del equipo investigador, para poner dicha información al alcance de la comunidad científica del laboratorio
4.3 Meta-análisis
En una segunda fase y a partir de la base de datos (que tendría metodología común) podrían generarse metaanálisis que darían a esta información una potencia estadística/evidencia científica indiscutible, para permitir hablar de PPORE desde de la ciencia y no desde los sentimientos o los intereses económicos.
No es lo mismo el tiempo de protrombina en población "normal" que en pacientes en tratamiento con anticoagulantes orales. Otros ejemplos sobre la diferencia entre diagnóstico y seguimiento se han indicado en los ejemplos anteriores
b Puede ser conveniente realizar la prueba en tiempos mayores, lo que puede permitirnos aceptar los especímenes en caso de retrasos imprevistos, por ejemplo, si el recorrido preanalítico es de dos horas, pero con frecuencia existen problemas de tráfico que pueden retrasar el trasporte, podemos diseñarlo con un tiempo de tres horas.
c Ej: si el bias para el analito X es 2.1 mg/dl para febrero, 2.0 para marzo y 2.05 para abril, podemos usar el dato, pero si los valores son 2.1, 12.7 y 8.5 sin una explicación lógica, probablemente tenemos un problema en la medición del analito más importante que el error que pueda generarnos la extracción periférica.

5.- Bibliografia
l.- Problemática de los puntos periféricos de obtención y recogida de especimenes Rev. Diag. Biol. 49:138-148 2000. Ruiz Falcó F, Sánchez Castiñeiras C, Martín Lluch E, Sanz Estebaran J., Sempere Molina C, Prieto Menchero S. [ Links ]
2.- Garantia de calidad de la fase preanalitica-extralaboratorio en puntos periféricos de obtención y recogida de especimenes. Sempere C., Bayo M., Enguix A, Galar GM, García Saavedra L, Llorca Escuín I, Martín Lluch JE, Prieto Menchero S, Ruiz Falcó F. Rev Diag Biol 1999; 48:140-150. [ Links ]
3.- NORMAS AEBM 98 :"Criterios generales de actuación y funcionamiento: competencia técnica y profesional de laboratorios de análisis clínicos para ofrecer un servicio de laboratorio efectivo". AEBM 1999. [ Links ]
4.- Muestras: del paciente al laboratorio. Güder WG, Narayanan S, Wisser H, Zawta B. BD PUBLICATIONS 1996. [ Links ]
5.- Catálogo de pruebas de los laboratorios clínicos. Manual de procedimientos. INSALUD. Madrid 1999.1SBN 84-3510311-0 (577 pags).N° de publicación INSALUD: 1.736 [ Links ]
6.- El papel de la Extracción Periférica en la Incertidumbre Analítica. Un Ejemplo: la glucosa en nuestro medio. Prieto Menchero S, García-Garzón A, Franquelo Gutierrez R. Poster al Vlll Congreso Nacional del Laboratorio Clínico. Sevilla 3-5 Junio 1999. [ Links ]
7.- Control Estadístico de Procesos Versión 0.75. 1999 JJ Prieto Tapia. http://junior.us.es/jnebrera/index.html [ Links ]
8.- Selection of medically useful quality-control procedures for individual tests done in a multitest analytical system . DD Koch, JJ Oryall, EF Quam, DE1 Feldbruegge, DE Dowd, PL Barry, and JO Westgard. Clin Chem 1990 36: 230-233. [ Links ]
9.- US Departament of Health and Human Services. Medicare, Medicaid and CLIA programs: Regulations implementing the Clinical Laboratory Improvement Amendments of 1998 (CLIA). Final Rule. Federal Register 1992;57:7002-7186. [ Links ]
10.- Hyltoft Petersen P, Ricos C, Stockl D, Liber JC, Baadenhuijsen H, Frazer C, Thienpont L. Proposed guidelines for the internal quality control of analytical results in the medical laboratory. Eur J Clin Chem Clin Biochem 1996; 34:983-999 [ Links ]
11.- Graphical interpretation of analytical data from comparison field method with a Reference Method by use of difference plots . Petersen PH, et al Clin Chem 43:11 2039-2046 (1997) [ Links ]
12.-Bland JM, Altman DG Statistical methods for assessing agrcement between two methods of clinical measuroment. Lancet 1986,i: 307-10 [ Links ]
13.- J.M. Domenech Massons. Métodos estadísticos en ciencias de la salud. Edit. Gráficas Signo. 1990 [ Links ]
14.- A.M. Padrós Fluvia et col. Estudio comparativo entre sueros y plasmas. Quim. Clin. 1994 ;13(1) 41-45 [ Links ]
15.- Measurement in Laboratory Medicine . P. Strike Butterworth 1996 [ Links ]
16 -C.G.Fraser. Interpretación de datos bioquímicos-clínicos. Ed Mayo 1988 [ Links ]
17.-System to monitor a portion of the total testing process in Medical Clinics and Laboratories: Evaluation of a SplitSpecimen Design. Shahangian S et al Clin Chem 45:2 269-280 (1999) [ Links ]
18.- Regresion-based reference limits:determination of sufficient sample size Virtanen A, et al Clin Chem 44:11 2353-8 (1998) [ Links ]
19.- Validity of lineal regression in method comparison studies: is it limited by the statistical model or the quality of the analytical input data? Stockl D, et al Clin Chem 44:11 2340-6 1998 [ Links ]
20.- Necessary sample size for method comparison studies based on regressión analysis Linnet K, Clin Chem 45:6 882-894 ( 1 999) [ Links ]
21.- M.l. Salve y C.Sempere. Fase premetrológica: efecto del transporte de especimenes de orina sobre los resultados. Quim. Clin. 1995 ;14(1):12-16 [ Links ]
22.- Stability of Common Analyles in Urine refrigerated for 24 h. before automated analysis by test strips Fromm P et al. Clin Chem 46:9 (2000):1384-6 [ Links ]
23.- Effect of serum-clot contact time on clinical chemistry laboratory results Zhang DJ et al Clin Chem 44:6 1325-33 1998 [ Links ]
24.- Prolonged Prothrombin Time and APTT due to underfilled specimen tubes with 109 mmol/L (3.2%) citrate anticoagulant Reneke J et al Am J Clin Pathol June 1998 745-757 [ Links ]
25.- Refrigerated storage improves the stability if the complete blood cell count and automated differential Wood BL, et al Am J Clin Pathol 1999;112:687-695 [ Links ]
26.- List of Analytes Preanalytical Variables. Güder WG, Narayanan S, Wisser 11, Zawta B.German Society of Clinical Chemistry 1996. [ Links ]

