










My SciELO
 Custom services
Custom servicesServices on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkActas Urológicas Españolas
Print version ISSN 0210-4806
Actas Urol Esp vol.27 n.10 Nov./Dec. 2003
NOTA CLÍNICA
TUMOR FIBROSO SOLITARIO. DOS CASOS ADICIONALES
CON IMPLICACIONES UROLÓGICAS
L. XAMBRE*, R. LAGES*, M. CERQUEIRA*, V. SILVA*, R. PRISCO, R. SANTOS,
F. CARREIRA*, M. HONAVAR**
*Servicio de Urología. **Servicio de Anatomía Patológica. Hospital Pedro Hispano. Matosinhos. Portugal.
TUMOR FIBROSO SOLITARIO. DOS CASOS ADICIONALES CON IMPLICACIONES UROLÓGICAS El término tumor fibroso solitario designa una entidad patológica bien definida. PALABRAS CLAVE: Tumor fibroso solitario. Mesotelioma fibroso. CD 34. Neoplasia pélvica. ABSTRACT SOLITARY FIBROUS TUMOUR. TWO ADDITIONAL CASE REPORTS WITH UROLOGICAL IMPLICATIONS Solitary fibrous tumour is a well defined pathological entity originally described as a tumour of the pleura, but the occurrence of this neoplasm has increasingly been described at other sites. At present the development of these tumours is recognized as possible in virtually all anatomical sites. The rarity of this type of pathology has not allowed up to the present time clarification of the histogenesis, clinical behaviour, treatment and prognosis of these tumours. KEY WORDS: Solitary fibrous tumours. Fibrous mesothelioma. CD 34. Pelvic neoplasms. |
Tumor fibroso solitario designa una neoplasia rara que con más frecuencia aparece en la pleura, visceral o parietal. Inicialmente descrito en 1931 y denominado mesotelioma fibroso localizado, esta lesión fue desde esa fecha denominada con diferentes términos como mesotelioma localizado benigno, submesotelioma, tumor fibroso localizado de la pleura, fibroma pleural, fibroma subseroso y fibroma solitario benigno.
Este enorme número de términos es sobre todo el resultado de diversas teorías procurando explicar su histogénesis. Evidencias histológicas, imunohistoquímicas y ultraestructurales han sido usadas para apoyar el origen mesotelial o mesenquimatoso de estas neoplasias, aunque la última sea actualmente la más aceptada. El reconocimiento del origen no mesotelial ayudó a eliminar la noción de que estarían confinadas a la cavidad torácica y motivó la descripción creciente de su aparecimiento con localizaciones extrapleurales tan dispares como en el hígado o senos perinasales. Sin embargo, el diagnóstico diferencial no es fácil y siguen siendo confundidos con otras entidades.
Su diagnóstico correcto es importante ya que en la mayor parte de las situaciones son neoplasias indolentes y plausibles de tratamiento quirúrgico, únicamente de exéresis.
CASOS CLÍNICOS
Caso 1
Paciente de sexo femenino, 77 años de edad, víctima de traumatismo del flanco/región lumbar derecha, por caída. Antecedentes de hipotiroidismo, fibrilación auricular, insuficiencia cardiaca e HTA, medicada con levotiroxina, digoxina, furosemida, disopiramida. Sin antecedente de patología urológica conocida o antecedentes quirúrgicos. Dio entrada en el Servicio Urgencia 24 horas después del episodio traumático por dolor lumbar derecho y hematuria macroscópica mantenida desde el traumatismo. Se presentaba hemodinámicamente estable, consciente, orientada. En el examen clínico era visible pequeña área de equimosis a nivel lumbar derecho. Abdomen blando, depresible, doloroso a la palpación profunda en flanco derecho/ hipocondrio derecho, sin señales de irritación peritoneal. De referir a nivel del hipogastrio presencia de masa de grandes dimensiones que al toque bimanual se presentaba como masa dura, contornos redondeados, ocupando prácticamente toda la pequeña bacía, no dolorosa y difícilmente mobilizable. Analíticamente presentaba Hb 10,1 g/dL, Leuc 12100/µL, Urea 54 mg/dL (N< 51,6 mg/dL), Creat 1,3 mg/dL (N<1,3 mg/dL), restantes parámetros bioquímicos sin alteraciones. Sometida a TAC abdominopélvico con contraste (Fig. 1) que evidenció hidroureteronefrosis bilateral, traumatismo renal derecho con laceración profunda del parénquima renal, afectando el excretor, con extravasamiento de contraste para el espacio perirrenal (grado IV de la clasificación de Moore). Sin evidencia de lesión de otras vísceras macizas o líquido libre intraperitoneal. Presencia de voluminosa masa pélvica, aparentemente dependiente del útero, irregular y heterogénea midiendo aproximadamente 10 por 12 cm, haciendo procedencia en la cara posterior de la vejiga, sin aparentemente invadirla.

FIGURA 1. TAC con contraste intravenoso (caso 1).
En vista de la estabilidad de los parámetros hemodinámicos, de los datos analíticos e imagiológicos, se optó por tratamiento conservador con monitorización de los parámetros vitales, analgesia, cobertura antibiótica con cefalosporina de 3ª generación, sueroterapia, reposo absoluto en cama, estudio de la neoformación pélvica y posterior derivación urinaria.
Se realizó RMN abdomino-pélvica (Fig. 2) que mostró masa heterogénea pareciendo corresponder al útero, muy heterogénea, predominantemente hipointensa en T2, presentando área central de licuefacción, compatible con voluminoso fibromioma/ conjunto de fibromiomas presentando degeneración quística central. Dilatación ureteral a lo largo de todo el trayecto abdomino-pélvico, estando los uréteres comprimidos por efecto de masa de la neoformación anteriormente descrita.

FIGURA 2. RMN pélvica (caso 1).
Con la tentativa de resolver el componente de uropatía obstructiva, posible factor de mantenimiento del extravasamiento, se intentó la colocación retrógrada endoscópica de catéteres doble J. La cistoscopia no reveló alteraciones de la integridad de la mucosa vesical, verificándose que toda la base vesical, trígono incluido, se encontraban elevados, dificultando el cateterismo, el cual sólo fue posible a la derecha, por no ser visible el meato ureteral izquierdo, no habiendo dificultad de progresión del catéter a la derecha. Teniendo en cuenta el crecimiento indolente de la neoformación y la inexistencia aparente de envolvimiento de estructuras vecinas, se procedió más tarde a exploración quirúrgica en el sentido de histerectomizar la paciente, una vez que los datos apuntaban para origen uterino y comportamiento "benigno" de la masa. Al contrario se constató presencia de la vejiga casi en la totalidad en posición suprapúbica, anteriormente empujada por voluminosa masa retroperitoneal, verificándose útero y parámetros de aspecto perfectamente normales. Ante este hecho, se procedió a biopsia con examen extemporáneo, el cual apenas reveló " neoplasia de células fusiformes", no garantizando información acerca de la benignidad/agresividad de la masa en causa. Se realizó entonces biopsia incisional de la masa, cateterismo ureteral "a cielo abierto" y colocación de cistostomía suprapúbica.
El examen histológico de la pieza de biopsia (Fig. 3) reveló neoplasia de células fusiformes, dispuestas en ejes irregulares, separadas por fibras de colágeno y con vasos de pared engrosada. Ausencia de atipia citológica significativa, pleomorfismo nuclear, necrosis, hemorragia o figuras de mitosis. El estudio inmunohistoquímico evidenció fuerte expresión de las células neoplásicas para vimentina y CD 34 (Fig. 3) y negatividad para actina, desmina, proteína S100, CAM 5.2, Caldesmon y CD 117. En conclusión, los aspectos morfológicos e inmunocitoquímicos coincidían con el diagnóstico de tumor fibroso solitario.
Disponiendo ya de diagnóstico de la neoformación, después de la revisión bibliográfica confirmando el comportamiento benigno de la mayor parte de estos tumores, se realizó la exéresis del tumor. El mismo se encontraba como ya fue referido en el plano entre la vejiga y la vagina/útero, no envolviendo ninguna de estas estructuras, una vez que la exéresis del mismo se hizo sin perjuicio de estas.
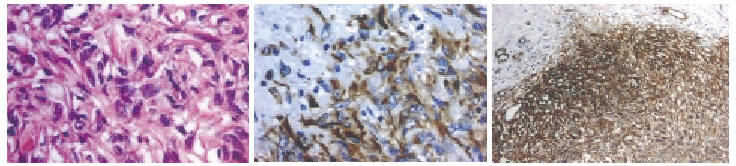
FIGURA 3. Tumor fibroso solitario - histologías. Hematoxilina-eosina, Vimentina, CD 34.
La pieza retirada (Fig. 4) estaba constituida por formación nodular de 657 g, superficie externa lobulada, no capsulada, presentando superficie de corte blanquecina, homogénea. Los aspectos de la microscopia y estudios inmunohistoquímicos reflejaban los ya descritos para la pieza de biopsia.
Después de 7 meses de seguimiento la paciente se encuentra asintomática, con resolución de las lesiones de orden traumática. Hasta el momento no hay evidencia de recidiva local o manifestaciones de enfermedad a distancia.

FIGURA 4. Fibroma solitario - pieza operatoria (caso 1).
Caso 2
Paciente de sexo masculino, 67 años de edad, antecedentes patológicos irrelevantes, que refiere historia de aparecimiento de tumefacción localizada al hemiescroto derecho, con cerca de un año de evolución, aumento de volumen progresivo y sin fluctuaciones, no dolorosa. Sin otras quejas. Negaba antecedentes compatibles con episodio de infección/inflamación escrotal. Sin historia de traumatismo escrotal o inguinal. Sin antecedentes quirúrgicos. Negaba igualmente antecedentes de tuberculosis.
Al examen físico era visible masa ocupando toda la bolsa escrotal derecha, aparentemente independiente del testículo, lisa, indolora, dura, movible con relación a los planos superficiales y profundos, presentando transiluminación negativa. No eran aparentes adenopatías inguinales homo o contralaterales. El examen abdominal no revelaba masas u organomegalias. Sin adenopatías palpables de otros territorios.
Realizada ecografía que puso en evidencia neoformación de patrón sólido con aproximadamente 10 cm de eje mayor, aparentemente dependiente del cordón espermático e independiente del testículo, heterogénea. El TAC abdomino-pélvico realizado a seguir corroboró estos datos. No eran visibles lesiones sospechosas de localización secundaria en los órganos macizos intra-abdominales. No eran igualmente visibles adenopatías, tanto en las cadenas pélvicas, como en las cadenas lumbo-aórticas.
Los parámetros analíticos hematológicos, el perfil bioquímico y los marcadores tumorales α-FP e β-HCG) no presentaban desvíos con relación a los valores de referencia.
Con estos datos, fue sometido a exploración quirúrgica por vía inguinal, donde se verificó que la neoformación se originaba a partir de las estructuras del cordón y fácilmente destacable de las estructuras vecinas. Se realizó examen extemporáneo que demostró neoplasia de células fusiformes, sin mitosis u otras características de agresividad.
Basados en este hecho y en la evolución clínica, fue realizada exéresis del tumor con margen quirúrgico, optándose por preservación de la gónada.
La histología definitiva corroboró los resultados del examen extemporáneo, demostrando neoplasia compuesta de células alargadas, de disposición aleatoria, sin marcadores de agresividad tales como mitosis, pleomorfismo nuclear, atipia citológica significativa o aspectos de necrosis. Tal como en el caso anterior, el estudio inmunohistoquímico evidenció positividad de las células neoplásicas para Vimentina y CD 34 y negatividad para los restantes marcadores, particularmente marcadores de células musculares, epiteliales o de las vainas nerviosas, permitiendo asignar el diagnóstico de tumor fibroso solitario (Tabla I).
TABLA I
CARACTERÍSTICAS DOS LOS CASOS PRESENTADOS

DISCUSIÓN
Los tumores fibrosos solitarios son neoplasias raras de la edad adulta, compuestas de células fusiformes, apareciendo más frecuentemente en la pleura, localización donde fueron inicialmente descritos. La primera descripción de los mismos se debe a Klemperer y Rabin1, en 1931. Estos autores dividieron las neoplasias de pleura en dos grupos distintos: difusas y localizadas. Pensaban que las primeras tenían origen "en la célula mesotelial totipotencial" y como tal se trataba de "verdaderos mesoteliomas". Las segundas se originaban "en el tejido areolar subpleural, presentaban crecimiento lento, alcanzaban grandes dimensiones antes de provocar signos y síntomas y eran sugestivas de fibromas, fibrosarcomas de bajo grado y eran factibles de escisión quirúrgica1". Desde la primera descripción, la histogénesis de estas lesiones ha sido objeto de gran controversia y debate. Varias teorías son defendidas para explicar la etiología/célula de origen de estas lesiones.
Desde entonces, se reconoció la posibilidad de aparecimiento de estas neoplasias presentando características histológicas, inmunohistoquímicas y ultra-estructurales indistinguibles en otras localizaciones, además de la pleura. Parece incluso que pocas localizaciones anatómicas escaparan de ser afectadas. Casos ocasionales envolviendo estructuras torácicas como el propio parénquima pulmonar2,3, mediastino4, pericardio y extratorácicas como la órbita, tejidos blandos del tronco y miembros, timo, tiroides, cavidad nasal o senos perinasales, hígado5,6 y concretamente estructuras urológicas como la vejiga7, próstata y vesículas seminales8,9 fueron descritos, comprobando la ubicuidad de estos tumores.
Estos casos vienen a dar razón a aquellos que defienden un origen diferente de la célula mesotelial para estos tumores, una vez que si en estructuras como el mediastino o peritoneo la hipótesis de origen a partir de superficies serosas o células submesoteliales parecía obvia, lo mismo no acontece en otros casos. Por otro lado, múltiples estudios muestran que las células que componen estos tumores no presentan las características inmunohistoquímicas o ultra-estructurales de la diferenciación mesotelial, sino al contrario características de diferenciación fibroblástica2,10. La hipótesis más consensual actualmente admite su origen en células dendríticas intersticiales CD 34 positivas, mesenquimatosas, presentes prácticamente en todos los departamentos orgánicos12, lo que está de acuerdo con los relatos anteriormente referidos de casos apareciendo en múltiples órganos distintos. Los casos aquí presentados reportan aparecimientos adicionales de tumores de este tipo localizados en estructuras extratorácicas, ambos con afectación de órganos urológicos.
Por lo que respecta a la epidemiología de los TFS (tumores fibrosos solitarios), las diversas series son concordantes en presentarlos como tumores de la edad adulta, con edad media de cerca de 56 años2,3,5,6,9 (existiendo sin embargo un caso descrito de una paciente con 17 años de edad con la misma localización anatómica del primer caso relatado por nosotros5). No parece existir predominancia por ninguno de los sexos. Estos datos demográficos no presentan diferencia significativa cuando comparamos tumores de localización pleural/subpleural12. La presentación clínica varía lógicamente con su localización. Característicamente los tumores pleurais o torácicos son descubiertos accidentalmente después de la realización de estudios de imagen con otros fines, en pacientes asintomáticos. Pueden sin embargo dar origen a tos o dispnea, hemoptisis o incluso a Síndrome de la Vena Cava Superior. Los TFS de la esfera urológica se manifiestan por sintomatología perfectamente inespecífica, como desconforto o sensación de masa pélvica, polaquiuria o retención urinaria por efecto compresivo sobre órganos próximos o hemospermia (origen en la vesícula seminal)9. El primer caso que describimos se reviste de características específicas. La paciente no presentaba ninguna manifestación clínica, tratándose de un hallazgo por imagen. Que sepamos, se trata de la primera descripción en el contexto de traumatismo renal. Si como ya es conocido, las situaciones de uropatía obstructiva predisponen a lesiones renales traumáticas, en ocasiones graves, con traumatismos poco intensos como en el caso 1, la causa de la uropatía obstructiva fue en este caso bastante rara. En el segundo caso, la manifestación fue únicamente la masa en sí misma y los síntomas locales que ocasionaba.
Están descritos casos raros de hipoglucemias graves (4% de los casos), en ocasiones incluso con coma hipoglucémico en pacientes con grandes masas tumorales de este tipo3,12,13 y asociación con fenómenos de osteoartropatía hipertrófica5 de difícil explicación. En nuestros casos no había en la historia de los pacientes ninguna evidencia de episodios o manifestaciones de este tipo.
El diagnóstico se basa en las características histológicas. En términos macroscópicos se presentan como tumores bien delimitados, superficie lisa, a veces multinodulares, no encapsulados pero fáciles de eliminar, generalmente alcanzando grandes dimensiones (existen incluso relatos de neoplasias de este tipo alcanzando varios Kg). Al corte presentan superficie blanca o cenicienta, homogénea o conteniendo áreas de hemorragia o necrosis (particularmente cuando son de grandes dimensiones). Estos mismos aspectos fueron verificados por nosotros. Los aspectos microscópicos revelan proliferaciones de células fusiformes, de núcleo alargado, cromatina dispersa y nucléolo ausente, rodeado de citoplasma escaso. Las células adoptan un patrón casual, fortuito ("paternless pattern" de los autores anglosajones) y están rodeadas y separadas entre sí por estroma colagenoso más o menos denso. De forma característica, áreas de gran densidad celular alternan con áreas hipocelulares. En la mayoría de las situaciones las lesiones muestran vascularización prominente, con numerosos vasos de pequeño y medio calibre, adoptando patrón de crecimiento hemangiopericítico. Relativamente a marcadores celulares, todos los autores son unánimes en afirmar que las células tumorales marcan fuertemente para el CD 34 (anticuerpo monoclonal contra el antígeno de la célula progenitora hematopoyética, anti HPCA-1) y para la Vimentina. Por otro lado, se revelan uniformemente negativos los estudios con otros anticuerpos monoclonales (citoqueratinas, desmina, actina, proteína S 100, colágeno tipo IV), lo que permite en la mayor parte de las situaciones realizar diagnóstico diferencial. Las situaciones que más problemas de diagnóstico diferencial colocan son schwannomas, neurofibromas y otros tumores benignos o malignos de las vainas nerviosas, sarcoma sinovial monofásico, leiomiosarcomas (diferenciables por los patrones histológicos y citológicos). La propensión para errores diagnósticos con esta entidad está bien documentada. Autores como Suster o Westra reportan errores en el diagnóstico inicial respectivamente en 91 y 80%6,9. El problema surge particularmente para tumores de localización extratorácica (aunque las características no sean diferentes de los demás) en los cuales el diagnóstico ni siquiera es considerado. Es en estas situaciones que el estudio inmunohistoquímico se reviste de mayor interés. Pese al hecho de la positividad para CD 34 no ser totalmente específica, cuando asociada a negatividad para otros marcadores y a los hallazgos de la microscopia óptica, permite distinguirlo de los diagnósticos diferenciales ya referidos con seguridad.
El comportamiento clínico de estos tumores es imprevisible. Aunque les sea atribuido un comportamiento mayoritariamente benigno, pueden sufrir recidivas (descritas en ocasiones muchos años después de la exéresis) e incluso metastatizaciones a distancia2,3,12. La presencia de este comportamiento agresivo fue estimada en cerca del 13 a 23% de las situaciones para los tumores torácicos y existen incluso autores que encuentran cifras mayores de este fenómeno para los tumores extratorácicos12-14. Por otro lado, la presencia/ ausencia de indicadores histológicos de agresividad como dimensión tumoral, celularidad elevada, pleomorfismo celular, presencia de número elevado de mitosis por campo de gran ampliación y la presencia de necrosis/hemorragia no son garantía de comportamiento indolente/agresivo. Sin embargo, en un análisis reciente (la mayor serie incluyendo tumores extratorácicos publicada hasta el momento, con "follow up" alargado), Gold et al.12 encontraron tasas de recidiva local y metastatización presentando diferencias significativas cuando consideraban tumores con más de 10 cm y presencia de un componente histológico maligno (que definieron como presencia de áreas de celularidad muy elevada - incluyendo más de 5% de la masa tumoral- y presencia de más de 4 mitosis por campo de gran ampliación). Estas diferencias eran particularmente marcadas cuando las dos condiciones están presentes simultáneamente12. En nuestros casos el único de estos factores presente es la gran dimensión de ambos tumores, haciendo preveer la necesidad de seguimiento atento.
El tratamiento recomendado, consensual, consiste en exéresis quirúrgica completa, no siendo necesarios actos quirúrgicos particularmente agresivos/alargados. Por otro lado, el uso de quimioterapia o esquemas de radioterapia neoadyuvantes o adyuvantes relatados apenas en casos puntuales, presentaron resultados dispares, no permitiendo sacar conclusiones válidas. De cualquier forma y como ya fue referido antes, el seguimiento a largo plazo de estos pacientes es fundamental, una vez que existen casos de recidiva local o a distancia muchos años después de la exéresis de estos tumores. Actualmente no disponemos de amplios seguimientos, aunque en ambos casos no se verifique por ahora ninguna evidencia de enfermedad.
REFERENCIAS
1. KLEMPERER P, RABIN CB.: Primary neoplasms of the pleura: a report of five cases. Arch Pathol 1931; 11: 385-412. [ Links ]
2. ENGLAND DM, HOCHHOLZER L, MC CARTHY MJ.: Localized benign and malignant tumours of the pleura. A clinicopathologic review of 223 cases. Am J Surg Pathol; 13: 640-658. [ Links ]
3. BRISELLI M, MARK EJ, DICKERSIN R.: Solitary fibrous tumour of the pleura: eight new cases and review of 360 cases in the literature. Cancer 1981; 47: 2678-2689. [ Links ]
4. YOUSEM SA, FLYNN SD.: Intrapulmonary localized fibrous tumour. Intraparenchymal so-called localized fibrous mesothelioma. Am J Clin Pathol 1988; 89: 365-369. [ Links ]
5. GOODLAD JR, FLETCHER CDM.: Solitary fibrous tumour arising at unusual sites: analysis of a series. Histopathology 1991; 19: 515-522. [ Links ]
6. SUSTER S, NASCIMENTO A, MIETTINEN M et al.: Solitary fibrous tumour of soft tissue: a clinicopathologic and immunohistochemical study of 12 cases. Am J Surg Pathol 1995; 19 (11): 1257-1266. [ Links ]
7. BAINBRIDGE TC, SINGH RR, MENTZEL T et al: Solitary fibrous tumour of the urinary bladder: report of two cases. Hum Pathol 1997; 28: 1204-1206. [ Links ]
8. NOGUCHI M, HIRABAYASHI Y, KATO S, NODA S.: Solitary fibrous tumour arising from the prostatic capsule. J Urol 2002; 168: 1490-1491. [ Links ]
9. WESTRA WH, GRENKO RT, EPSTEIN J.: Solitary fibrous tumour of the lower urogenital tract: a report of five cases involving the seminal vesicles, urinary bladder and prostate. Hum Pathol 2000; 31 (1): 63-68. [ Links ]
10. DERVAN PA, TOBIN B, OCONNOR M.: Solitary (localized) fibrous mesothelioma: evidence against mesothelial cell origin. Histopathology 1986; 10: 867-875. [ Links ]
11. VAN DE RIJN M, LOMBARD CM, ROUSE RV.: Expression of CD 34 by solitary fibrous tumour of the pleura, mediastinum and lung. Am J Surg Pathol 1994; 18: 814-820. [ Links ]
12. GOLD JS, ANTONESCU CR, HADJU C, COIT DG et al.: Clinicopathologic correlates of solitary fibrous tumours. Cancer; 94 (4): 1057-1068. [ Links ]
13. NELSON B, BURMAN SO, KIANI R et al.: Hypoglicemic coma associated with benign pleural mesothelioma. J Thorac Cardiovasc Surg 1975; 69: 306-314. [ Links ]
14. VALLAT-DECOUVELAERE AV, DRY SM, FLETCHER CD.: Atypical and malignant solitary fibrous tumours in extrathoracic locations: evidence of their comparability to intra-thoracic tumours. Am J Surg Pathol 1998; 22: 1501-1511. [ Links ]
Dr. L. Xambre
Servicio de Urología. Hospital Pedro Hispano
Rua Dr. Eduardo Torres
4464-509 Matosinhos (Portugal)
(Trabajo recibido el 28 enero 2003)

