










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActas Urológicas Españolas
versión impresa ISSN 0210-4806
Actas Urol Esp vol.30 no.1 ene. 2006
Desarrollo de un microarray tisular (TMA) para el estudio inmunohistoquímico del patrón de expresión molecular en el cáncer de próstata (Parte 1)
Development of Tissue Microarray Technology (TMA) for immunohistochemical study of molecular expression profiling in prostate cancer (Part 1)
J.A. Lorente Garin1,3, J. Lloreta Trull2,3, C. Allepuz Losa4, L. Plaza Mas5, L.A. Rioja Sanz4, A. Gelabert Mas1,3
1Servicio y Cátedra de Urología, 2Servicio de Anatomía Patológica, 3Laboratorio de Uro-Patología Aplicada, Hospital del Mar, Barcelona. 4Servicio y Cátedra de Urología, 5Servicio de Anatomía Patológica, Hospital Universitario Miguel Servet, Zaragoza.
Estudio becado por la Fundación para la Investigación en Urología (FIU) de la Asociación Española de Urología.
Dirección para correspondencia
RESUMEN
El microarray tisular (TMA) es considerado hoy en día una potente herramienta para el análisis masivo del perfil molecular del cáncer. En este trabajo describimos la experiencia de ambos centros en el diseño y creación de un TMA para el estudio de la expresión molecular del cáncer de próstata así como una revisión de los diferentes aspectos técnicos necesarios para el análisis de los resultados obtenidos con esta técnica. En la actualidad, se están estudiando los datos generados por las distintas técnicas inmunohistoquímicas para la obtención de un patrón molecular en diferentes estadios clínicos.
Palabras clave: Microarray tisular. Cáncer de próstata. Inmunohistoquímica
ABSTRACT
Tissue microarray technology (TMA) is nowadays considered as a powerful tool for the high-throughput analysis of molecular expression pattern of cancer. In this manuscript we show the experience of both groups in the design and building of a TMA for the study of protein expression pattern of prostate cancer as well as a summary of the technical points to analyze the results obtained with this technology. Today, different data generated by the immunostained tissues are studied to achieve a molecular profile in different clinical scenarios.
Key words: Tissue microarray. Prostate cancer. Immunohistochemistry.
La finalización del Proyecto del Genoma Humano, ha permitido nuevas oportunidades de análisis de genes y sus productos en el tejido humano. Es de esperar que en los próximos años se establezcan nuevas clasificaciones moleculares que aportarán información relevante para la tipificación de cada tumor. El mayor conocimiento de los mecanismos moleculares implicados en la génesis y progresión del cáncer es, por lo tanto, la base para la creación de nuevas herramientas para predecir el potencial maligno de los diferentes tumores. Estas nuevas técnicas tienen en común la capacidad de generar con fiabilidad y rendimiento una gran cantidad de datos emergentes que, por otra parte, sería imposible obtener por los métodos tradicionales de la patología molecular. Para la creación de estos futuros mapas o patrones moleculares, es de gran utilidad a nivel clínico el empleo de la técnica del microarray tisular (TMA), al permitir la realización de diferentes ensayos en cientos de muestras simultáneamente. Desde su desarrollo ha demostrado ser un potente instrumento para la investigación, que ofrece la oportunidad de plantear importantes estudios retrospectivos de carácter multicéntrico y se ha convertido en el eje central de ejemplares colaboraciones en el estudio del cáncer de próstata, como el Cooperative Prostate Cancer Tissue Resource del National Cancer Institute1 o el Prostate Specialized Program of Research Excellence Model2 de las Universidades de Michigan y Johns Hopkins. Con este trabajo, pretendemos comunicar nuestra experiencia en el diseño y creación de un TMA para el estudio de la expresión inmunohistoquímica de los procesos implicados en la biología del cáncer de próstata, así como, revisar los diferentes aspectos técnicos necesarios para el análisis de los resultados obtenidos con esta técnica de creciente implantación.
Microarray tisular
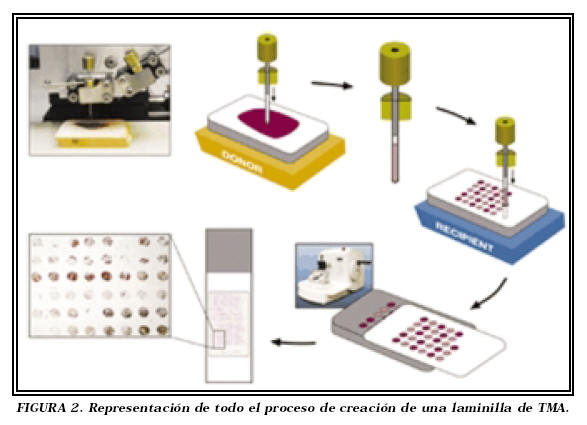
Los TMAs proporcionan un nuevo método de alto rendimiento para estudiar el perfil molecular, la expresión del mRNA o el análisis de las alteraciones cromosómicas mediante hibridación in situ de múltiples muestras de tejido simultáneamente3. Esta técnica proporciona la posibilidad de estudiar nuevos marcadores o los ya existentes de manera masiva a la vez que facilita las colaboraciones intra-e interinstitucionales. Estos chips de tejido pueden facilitar y acelerar el traslado de la información obtenida a la práctica clínica ya que obvian las limitaciones de las técnicas convencionales, como la necesidad de estudiar uno a uno miles de especímenes para su estudio repitiendo los ensayos y con las consiguientes y extensas validaciones. A su vez, el TMA preserva las muestras originales de tejido que con los métodos tradicionales disminuyen en cuantía y se deterioran por el uso frecuente. El microarray de tejido (o micromatriz tisular) se construye adquiriendo muestras cilíndricas de cientos de tumores o zonas de interés a partir de los bloques de parafina. Todos los cilindros así obtenidos se incorporan a un bloque de parafina receptor que puede contener hasta 600 muestras y que es a su vez cortado en múltiples secciones para llevar a cabo ensayos mediante técnicas inmunohistoquímicas o de hibridación in situ fluorescente. Un único ensayo o reacción de hibridación in situ proporciona así información de todos los especímenes de la laminilla a la vez, reproduciéndose fidedignamente las condiciones del ensayo para todas las muestras. La construcción de bloques repetidos puede permitir múltiples secciones de los mismos tumores para su estudio. Las zonas de especial interés para biopsiar en la pieza donante son analizadas y marcadas de forma convencional en tinciones de H-E. De cada bloque pueden ser tomadas muestras de 0,6 ó 1 mm. de diámetro y 3-4 mm. de longitud mediante un arrayer (Fig. 1) e incluidas a modo de matriz en un nuevo bloque receptor de parafina. De este bloque se realizan secciones de 6-8 micras para cada laminilla (Fig. 2).

Material y métodos
Inclusión de pacientes para TMA
El primer paso en el diseño de un TMA típico es la selección de los pacientes a estudiar. La posibilidad intrínseca a la técnica de analizar muchos casos, permite la inclusión de un número suficiente de pacientes en diferentes situaciones clínicas. Para ello es de gran utilidad el trabajo con bases de datos clínicas y patológicas actualizadas. En nuestro estudio se seleccionaron 227 pacientes, procedentes de las bases de datos de prostatectomías radicales del Hospital del Mar (127) y del Hospital Universitario Miguel Servet de Zaragoza (100). Se realizó una selección previa de los casos atendiendo a los criterios clínicos para cirugía radical en los dos centros, la calidad del material disponible, el tiempo de seguimiento de los pacientes y la posibilidad de tratamiento hormonal previo a la prostatectomía. De esta manera, se obtuvo información relevante para crear una base de datos conjunta con una serie homogénea de casos, 99 pacientes sometidos a diferentes tiempos de bloqueo neoadyuvante y 128 no tratados con diferentes tiempos de seguimiento (entre 12 y 120 meses). Esta revisión fue realizada con la aprobación del Comité de Bioética del IMAS.
Variables clínicas y patológicas. Bases de datos
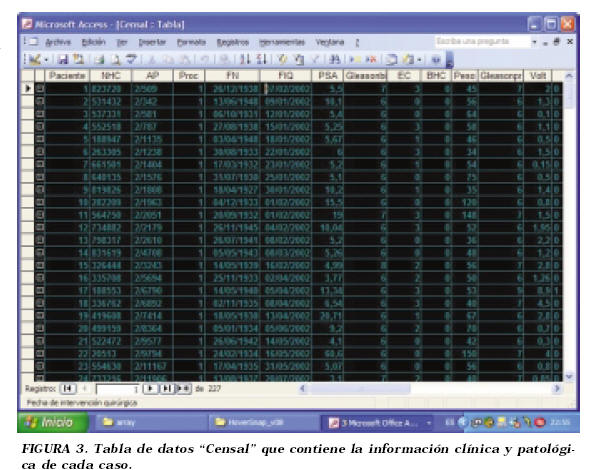
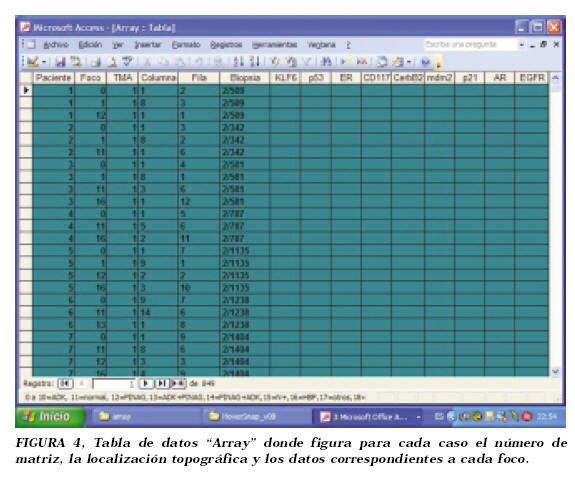
La información clínica y patológica obtenida fue introducida en una base de datos relacional (TMA DB) creada al efecto en Microsoft Access 2002 (Microsoft, Redmond,WA) para organizar y relacionar todos los niveles de información a medida que fueran creándose los TMAs y realizándose las diferentes lecturas de las técnicas de inmunohistoquímica. Este tipo de solución evita el tener que introducir los datos de manera redundante, como se haría en una hoja de cálculo. El TMA DB se compone de una tabla de datos Censal (Fig. 3) que contiene la información clínica y patológica del paciente con clave principal autogenerada y vinculada a otras dos tablas de datos: una llamada Array donde figura para cada caso la fila (Y) y columna (X) que le corresponde en su matriz junto con los datos del foco incluido (Fig. 4), y en la cual se irán incorporando los datos de las distintas técnicas inmunohistoquímicas para cada foco, generadas por la lectura manual de las tinciones. La tercera tabla es la llamada Ariol image creada con los datos de lectura automatizada a través de un sistema de cuantificación mediante digitalización de la imagen. Para prevenir entradas duplicadas para los datos de cualquier paciente o información duplicada de los focos, se han generado máscaras de entrada que emiten mensajes de error. Estos son algunos de los ejemplos de las precauciones observadas para intentar minimizar los errores en la entrada de los datos. Para facilitar los posteriores análisis estadísticos, todos los valores han sido introducidos como variables numéricas, evitando en la medida de lo posible el texto (Tabla 1).

Construcción del TMA
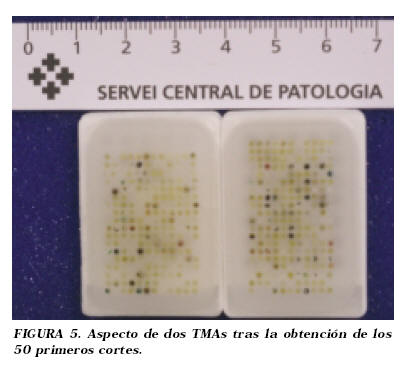
El primer paso en la construcción de un TMA es el análisis y preparación de los bloques donantes. Se toman laminillas para tinción con hematoxilina y eosina (H&E) de referencia en todos los bloques para revisar los diagnósticos y marcar con tinta las zonas de interés. Un tema frecuentemente repetido es si las pequeñas biopsias empleadas en la construcción de un TMA ofrecen información suficientemente representativa de lo que es un tumor mucho mayor. Debe tenerse en cuenta, que el principio básico para un análisis con TMA es fundamentalmente diferente al de un estudio histológico convencional. Se trata de una técnica de investigación a nivel poblacional y no debe ser considerada como una técnica diagnóstica de casos individuales. Esto es especialmente relevante en el estudio del cáncer de próstata, donde generalmente el tumor es multifocal y heterogéneo. Estas limitaciones se han obviado con un análisis minucioso de los bloques donantes y una toma selectiva y precisa de las múltiples zonas de interés, a la vez que aumentando el muestreo de cada tipo de lesión en cada caso. Estas medidas han demostrado ser más eficaces a la hora de capturar la heterogeneidad del tumor que el empleo de biopsias de mayor tamaño4. En nuestro estudio, dada la heterogeneidad del tejido y la posibilidad de tomar muestras poco representativas del tumor, este proceso fue evaluado por duplicado por los patólogos de ambos centros, consensuando criterios como el grado histológico y la localización de las zonas a incluir. Se consideraron como muestras de interés los focos múltiples de tumor, la neoplasia intraepitelial prostática de alto grado (PINAG) adyacente o no a la neoplasia, la hiperplasia benigna (HBP), la próstata normal y los ganglios linfáticos. En total obtuvimos 849 focos de estudio (3,74 focos/paciente, rango 2-10) pertenecientes a 564 focos con cáncer de próstata, 146 focos con PINAG (66 aislado y 80 adyacente a tumor), 73 focos con HBP, 60 focos considerados próstata periférica normal y 6 focos con metástasis ganglionares. De los diferentes bloques donantes, y una vez localizados los focos a adquirir, se obtuvieron mediante un arrayer manual (Beecher Instruments, Silver Spring, MD, USA) las biopsias de 0,6 mm a integrar en el bloque receptor. De esta manera se construyeron 4 TMAs. Las muestras fueron introducidas aleatoriamente en los bloques, pero siguiendo una distribución lineal con una distancia de 1 mm entre los centros de cada muestra, dando lugar a dos tipos de matrices: de 18 columnas por 12 filas y de 20 columnas por 11 filas (Fig. 5). Una vez que los bloques receptores estuvieron construidos, se seccionaron en láminas de 6 micras para realizar un control de calidad con tinción de H&E. En nuestra experiencia, fue posible obtener una media de 50 laminillas por bloque sin pérdida significativa del número de cilindros en los últimos cortes, por lo que el número de focos potenciales para estudio es de 42.450. Se emplearon como casos control 5 biopsias en cada TMA de tejido procedente de vejiga urinaria y de amígdala. En cada TMA los controles fueron colocados en diferentes localizaciones. Todo el proceso fue representado manualmente a medida que se construía en unas plantillas para facilitar el paso de los datos a la TMA DB.
Tinciones inmunohistoquímicas
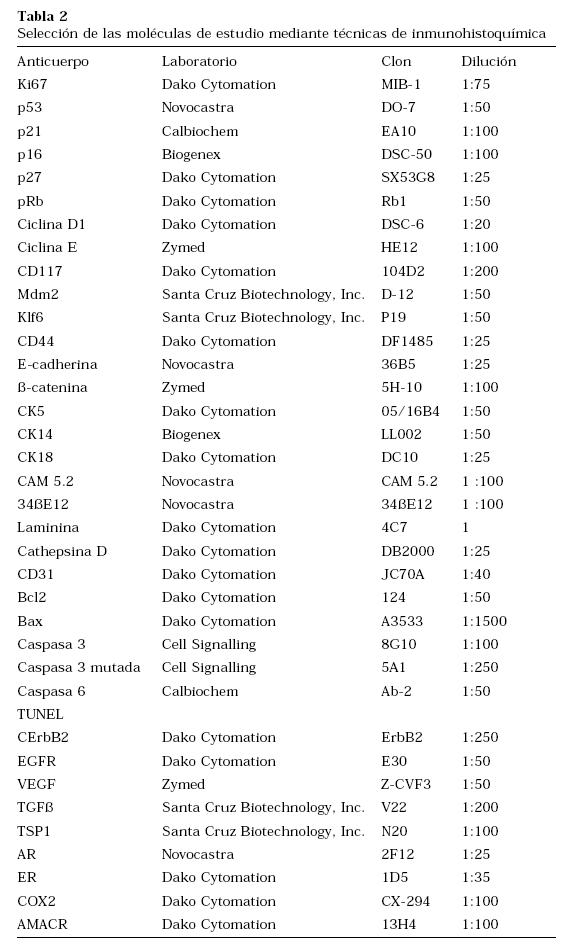
Se ha postulado, que la sobreexpresión relativa de moléculas relacionadas con la capacidad de invasión y la reducción de las relacionadas con la adhesión, es un buen predictor del estadio patológico en el cáncer de próstata5. La mayoría de estos estudios, sin embargo, se centran en aspectos individuales de la progresión tumoral, debido a la dificultad para confeccionar un patrón de expresión molecular mediante técnicas convencionales. En este sentido y conocida la ventaja que confiere el TMA, consideramos de gran interés realizar un estudio que integre los diferentes procesos implicados en la biología tumoral, analizando la interrelación existente en la expresión proteica diferencial y su posible utilidad en la predicción del estadio patológico. Se ha considerado el análisis conjunto de las moléculas implicadas en procesos básicos, como la adhesión celular y la degradación del estroma, al igual que la expresión de proteínas relacionadas con el control del ciclo celular, la proliferación y la apoptosis. Dadas las características del cáncer de próstata, se ha considerado importante analizar la expresión de receptores hormonales, moléculas implicadas en la angiogénesis y su relación con los factores de crecimiento (growth factors). Con la elección de un total de 36 moléculas a estudiar mediante técnicas de inmunohistoquímica, pensamos que se analizan la mayoría de los procesos biológicos básicos y ello puede permitir la confección de un patrón de expresión molecular suficientemente representativo (Tabla 2). Si se demostrara un patrón uniforme con capacidad predictiva, sería de gran interés su validación en la biopsia previa y, posteriormente, su validación prospectiva en nuevos pacientes.
El análisis de distintas zonas de cada tumor merece especial atención. Se ha demostrado la existencia de una expresión diferencial y heterogénea dentro de cada tumor. Las zonas tumorales más periféricas y/o con extensión extraprostática y perineural parecen tener mayor expresión de moléculas de digestión estromal y menor de adhesión celular, expresión de un mayor potencial de diseminación e invasión5. Se consideró por este hecho el estudio individual de la zona tumoral central, periférica y de márgenes positivos o extraprostáticos al igual que los ganglios linfáticos metastásicos caso de haberlos.
Adquisición automatizada de la Imagen y Análisis usando el Ariol SL-50
Además del análisis visual, los niveles de expresión inmunohistoquímica en un subgrupo de arrays se evalúan siguiendo una lectura automatizada a través de un sistema de cuantificación mediante digitalización de la imagen (Ariol SL-50, Applied Imaging, Corp.,Santa Clara,CA) (Fig. 6). El Ariol SL-50 consiste en un microscopio Olympus Bx 61 automatizado acoplado a una videocámara CCD de alto rendimiento capaz de detectar niveles de matiz, saturación y luminosidad. El brazo neumático robotizado es capaz de cargar 50 laminillas. Aplicando el software correspondiente, el umbral de cada parámetro es optimizado para traducirse en intensidad de colores. El software con el que va equipado, además de otras aplicaciones, contiene tres módulos para la interpretación de las imágenes escaneadas en función de la localización de la proteína identificada por la técnica inmunohistoquímica, Hersight para análisis y cuantificación de proteínas de membrana, Kisight para marcadores nucleares y Aesight para proteínas citoplasmáticas (Fig. 7). Los datos ofrecidos por esta lectura automatizada en forma de intensidad, porcentaje y distribución son almacenados para su posterior tratamiento como variables cuantitativas en la tabla de datos Ariol image.


Análisis estadístico de los datos procedentes de un TMA
Uno de los retos más importantes con el que nos enfrentamos en estas técnicas de alto rendimiento, es la necesidad de manejar efectivamente la gran cantidad de datos generados. Las soluciones propuestas van desde asociar una combinación de herramientas convencionales, hasta el desarrollo de nuevas aplicaciones específicas para este propósito. Una demostración on-line de esta alternativa está disponible en http://genome-www.stanford.edu/TMA/explore.shtml. Lo recomendable es escoger para el almacenamiento un formato que permita la exportación fácil de los datos y la posibilidad de emplear para el tratamiento estadístico técnicas que busquen relaciones entre los resultados de las diferentes tinciones inmunohistoquímicas o agrupaciones de las observaciones según su similitud, como el análisis de la asociación de los grupos no supervisado (clustering analysis), supervisado (árboles de decisión) o las diferentes técnicas de regresión. Una situación habitual es que las covariables estén dispersas, no normalizadas y altamente relacionadas entre sí. Los investigadores que trabajan con TMA de forma rutinaria dicotomizan los valores de los marcadores. Sin embargo, el empleo de un sistema mixto con lectura automatizada permite cuantificar los valores del marcador, requiriendo de estudios de correlación. Se han revisado diferentes métodos para evaluar el valor predictivo de los modelos de regresión de Cox y para demostrar si los datos ofrecidos por el TMA contienen este tipo de información6. Asimismo, en la literatura se ha comunicado el empleo de métodos no paramétricos de replicación bootstrap para validar el índice de concordancia. Estos métodos pueden utilizarse como herramienta para estimar el grado de significación estadística de la contribución de diferentes covariables durante la construcción de un modelo. Esto es especialmente importante cuando se trabaja con poca representación tumoral y es uno de los motivos por los que existen varias muestras de un mismo tumor. No obstante, se debe tener en cuenta que una parte de la variabilidad puede no estar explicada por una verdadera expresión diferencial, sino por la variabilidad que introducen los distintos elementos que intervienen en todo el proceso de obtención de los resultados. Diferentes métodos de análisis pueden tener una profunda influencia en la interpretación de los resultados, por lo que es absolutamente imprescindible adaptar el diseño del estudio a los consejos de expertos en sistemas bioinformáticos7.
Conclusión
El TMA demuestra ser un potente instrumento para la investigación ayudando al análisis masivo del perfil molecular de la patología tumoral. Facilita y acelera el traslado de la información obtenida a la práctica clínica, obviando muchas de las limitaciones de las técnicas convencionales. Esto permitirá en un próximo futuro tipificar cada tumor ofreciendo un tratamiento más individualizado y racional. Para rentabilizar todo su potencial se impone que los estudios basados en TMA sean multidisciplinares y multicéntricos, con revisión meticulosa de los casos, captura de los datos clínicos de bases de datos actualizadas, construcción meticulosa del array y un oportuno asesoramiento computacional y bioinformático. Esto permite aunar esfuerzos, conocimientos y experiencias con el fin de lograr la consecución del objetivo de forma más rápida y exitosa.
Referencias
1. Berman JJ, Datta M, Kajdacsy-Balla A, Melamed J, Orenstein J, Dobbin K, et al. The tissue microarray data Exchange specification: implementation by the Cooperative Prostate Cancer Tissue Resource. BMC Bioinformatics 2004;5:19. [ Links ]
2. Manley S, Mucci NR, De Marzo AM, Rubin MA. Relational database structure to manage high-density tissue microarray data and images for pathology studies focusing on clinical outcome. The prostate specialized program of research excellence model.Am J Pathol 2001;159:837-843. [ Links ]
3. Kononen J, Bubendorf l, Kallioniemi A, Barlund M, Schraml P, Leighton S, et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med 1998;4:844-847. [ Links ]
4. Kallioniemi OP, Wagner U, Kononen J, Sauter G. Tissue microarray technology for high-throughput molecular profiling of cance. Human Mol Genet 2001;10:657-662. [ Links ]
5. Kuniyasu H, Troncoso P, Johnston D, Bucana CD, Tahara E, Fidler IJ, et al. Relative expression of type IV collagenase, E-cadherin, and vascular endothelial growth factor/vascular permeability factor in prostatectomy specimens distinguishes organ-confined from pathologically advanced prostate cancers. Clin Cancer Res. 2000;6:2295-2308. [ Links ]
6. Liu X, Minin V, Huang Y, Seligson DB, Horvath S. Statistical methods for analyzing tissue microarray data. J Biopharm Stat 2004;14:671-685 [ Links ]
7. Quakenbush J. Computational analysis of microarray data. Nature Reviews Genetics 2001;2:418-427. [ Links ]
![]() Dirección para correspondencia:
Dirección para correspondencia:
Dr. J.A. Lorente Garín
Servicio de Urología.
Hospital del Mar
Passeig Maritim, 25-29
08003 Barcelona
E-mail: jlorente@imas.imim.es
Trabajo recibido el 24 de febrero 2005

